




Chiba Medical J. 95E:71-77, 2019
doi:10.20776/S03035476-95E-5-P71
[ Chiba Medical Society Award Review ]
Arifumi Iwata
Department of Allergy and Clinical Immunology, Graduate School of Medicine, Chiba University, Chiba 260-8670.
(Received September 2, 2019, Accepted September 12, 2019, Published October 10, 2019.)
For an adequate response to several types of invasive pathogens, CD4+ T cells differentiate intohelper T cell subsets with a specific function and mediate sequential immune responses. Antigen dose and affinity, as well as cytokines derived from innate immune cells, determine T cell fates including their diverse gene expression profiles and Th subset differentiation; however, the precise molecular mechanisms underlying such processes are still unknown. Interferon regulatory factor 4 (IRF4) and basic leucine zipper transcription factor, ATF-Like (BATF) , are expressed in hematopoietic cells. IRF4 and BATF form complexes that play pivotal roles in Th2, Th9, Th17, and Tfh cell differentiation via the regulation of chromatin accessibility, the recruitment of other transcription factors, and the induction of downstream genes including master regulators. Moreover, a recent study revealed that IRF4-BATF complexes act as the sensor for TCR signal strength and tune the diverse transcriptional program. This review highlights the pivotal roles of IRF4 and BATF complexes in T cell fate and their molecular mechanisms.
IRF4-BATF complex, Th subset differentiation, TCR signal strength, epigenome, pioneer factor
An adequate adaptive immune response is essential to maintain homeostasis, modulate the microbiome, clear invasive foreign pathogens, and prevent cancer development, allergy, and autoimmunity. An insufficient immune response leads to uncontrolled pathogen invasion and cancer development. Conversely, an excessive immune response causes host damage, allergy, and autoimmunity. Thus, an appropriate CD4+ helper T cell (Th cell) response including specific cytokine production, the number of expanding cells, and the timing of inflammation resolution must be finely tuned. However, how these T cell fates are appropriately regulated is largely unknown[1]
Th cells play central roles in adaptive immunity. When naïve CD4+ T cells recognize antigens on the major histocompatibility complex (MHC) class II via the T cell receptor (TCR) , they proliferate and differentiate into specific Th cell subsets[ Th1, Th2, Th9, Th17, follicular helper T (Tfh) , and regulatory T (Treg) cells]. These cells are characterized by the expression of specific transcription factors (TFs) , which are called as“ master regulators”, the production of a series of cytokines, and effector functions[2]. Antigen amounts and affinities for the TCR create a gradient of TCR signal strengths, which controls diverse T cell fates such as levels of expansion, activation status including exhaustion, various gene expression profiles, and Thsubset differentiation[3]. Recent studies have elucidated the importance of two TFs, interferon regulatory factor 4 (IRF4) and basic leucine zipper transcription factor, ATF-Like (BATF) , in Th subset differentiation[4,5], as well as the diverse outcomes derived from graded TCR strength[6,7]. This review highlights the crucial role of IRF4 and BATF complexes in T cell fates, in addition to the underlying molecular mechanisms.
IRF4
IRF4 is a member of the interferon regulatory factor (IRF) family. It has two domains, a DNA binding domain (DBD) and an IRF association domain, which are separated by a flexible linker. DBDs of all IRFs recognize and bind the core sequence GAAA, but IRF4 has only weak binding affinity. For stable transcriptional activity, IRF4 requires interactions with other TFs such as PU.1, SPI-B, BATF, signal transducer and activator of transcription (STATs) , nuclear factor of activated T cells (NFATs) , RAR-related orphan receptor-γt (ROR-γt) , SMADs, B-cell lymphoma 6 (BCL-6) , and forkhead box P3 (Foxp3)[4]. IRF4 expression is restricted to hematopoietic cells and unlike other IRFs, its expression is not induced by interferon, but rather by other factors such as IL-4, lipopolysaccharide, and TCR signals[4]. IRF4 regulates class switch recombination in B cells, plasma cell development, Th subset differentiation, the expansion and effector function of CD8+ T cells, and the development of cDC2s, which are also known as IRF4- dependent dendritic cells (DCs)[8]
BATF
BATF is a basic leucine zipper (bZIP) transcription factor of the activator protein-1 (AP1) family. BATF, BATF2, and BATF3 comprise the BATF subfamily. BATF has a bZIP domain, which contains a basic DNAbinding region and a leucine zipper region, but no transactivation domain. All BATF subfamily members heterodimerize with JUN family proteins such as c-JUN, JUNB, and JUND. BATF-JUN heterodimers recognize and bind TPA response elements with TGA (C/G) TCA motifs. BATF expression is also restricted to hematopoietic cells such as T cells, B cells, and DCs. BATF is induced by stimulation with IL-1, IL-4, IL- 6, GM-CSF, and TCR signals and plays crucial roles in class switch recombination in B cells, Th subset differentiation, and the effector function of CD8 T cells[5]. BATF3, a member of the BATF subfamily, is predominantly expressed in DCs and their progenitors and is also expressed in activated T cells. BATF3 compensates for the function of BATF. Therefore, the roles of BATF have been underestimated in Batf-/- mice[5,6]
Formation of the IRF4- BATF complex
In B cells and DCs, IRF4 mainly interacts with PU.1 or SPI-B and binds Ets-interferon composite elements (EICEs) with GGAAnnGAAA motifs (‘n’ indicates any nucleotide) . However, IRF4 in T cells mainly interacts with BATF, due to the low expression of PU.1 and SPI-B, and binds AP-1-interferon composite elements (AICEs) such as AICE1[ TTTCnnnnTGA (C/G) TCA] and AICE2[ GAAATGA (C/G) TCA][9,10]. IRF4 binding capacity in T cells completely depends on interactions with BATF because IRF4 in Batf-/- T cells cannot bind genomic DNA. Similarly, BATF binding is significantly reduced in Irf4-/- T cells[6,9,10,11]. Therefore, the formation of IRF4- BATF-DNA complexes is critical for stable binding and transcriptional activity.
The mucosal barrier and innate immune cells, upon encountering various pathogens and stimuli, establish an appropriate microenvironment via the production of a series of cytokines. This environment induces the expression of lineage-specific TFs and master regulators in activated CD4+ T cells and promotes Th subset differentiation, which is important for sequential immune responses (Fig.1)[2]
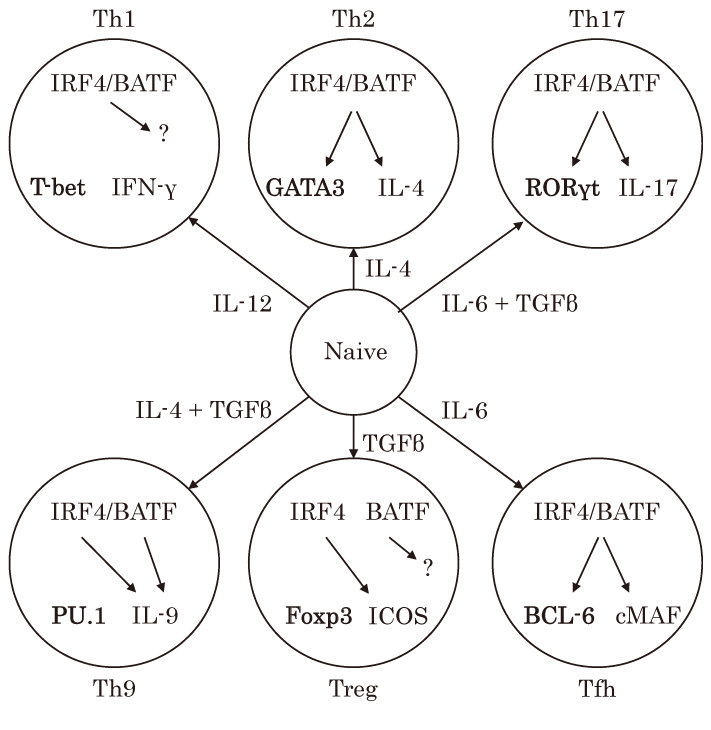
Fig.1 Th subsets and the IRF-BATF complex
When naïve CD4+ T cells are activated, microenvironmentderived cytokines induce the differentiation of naïve CD4+ T cells to distinct Th subsets. Six different Th subsets are characterized by the expression of master regulators (depicted as bold) and effector molecules. IRF4 and BATF regulate some master regulators and effector molecules cooperatively or independently.
Th1 cells
IL-12, produced by antigen-presenting cells, induces activation of the lineage-specific TF STAT4 in T cells. Activated T cells express the master regulator T-bet, produce interferon-gamma (IFN-γ) , and differentiate into Th1 cells, which protect the host from intracellular bacteria and virus infection. Both Irf4-/- and Batf/ Batf3 -double deficient CD4+ T cells normally express IFN-γ under Th1 conditions in vitro. Therefore, T-bet and IFN-γ in Th1 cells are not regulated by IRF4 or BATF[4,12]
Th2 cells
Th2 cells are induced by IL-4, activate STAT6, express GATA-binding protein 3 (GATA3) , produce type 2 cytokines such as IL-4, IL-5, and IL-13, and play critical roles in helminth expulsion and allergy. Irf4-/- CD4+ T cells fail to develop into Th2 cells in vitro and in vivo[4]. Similarly, Batf/Batf3 -double deficient T cells stimulated with Th2 culture conditions fail to produce type 2 cytokines[6,12]. ChIP-seq analysis, reported by several groups, showed that both IRF4 and BATF bind the promoter and enhancer around key genes of Th2 cell such as Gata3 , growth factor independent 1, Il4 , Il10 , and cytotoxic T-lymphocyte-associated protein 4 (Ctla4) . Indeed, in a murine model of asthma, Batf-/- mice show reduced asthmatic features including eosinophil infiltration, airway hyperresponsiveness, and type 2 cytokine production in the lung[13]. Therefore, IRF4 and BATF play indispensable roles in Th2 cell differentiation. Recently, it was shown that BTB and CNC homology, basic leucine zipper transcription factor 2 (BACH2) , which is a well-known negative regulator of effector differentiation, physically interacts with BATF. BATF-BACH2 complexes suppress Th2 differentiation by interfering with the formation of IRF4-BATF complexes on AICEs[14]
Th9 cells
Th9 cells develop from naïve CD4 T cells stimulated by IL-4 and transforming growth factor-β (TGF- β) . These cytokines activate STAT6 and SMAD2/3, induce PU.1 and IRF4, and induce the production of IL- 9. Th9 cells play a pathogenic role in allergic diseases[4]. IRF4 and BATF cooperatively bind the Il9 gene and induce IL-9 production; moreover, both Irf4-/- and Batf-/- T cells exhibit reduced IL-9 production under Th9 conditions. However, the regulation of Th9 differentiation by IRF4 and BATF is very complicated. PU.1 is highly expressed in Th9 cells, and thus, IRF4 might function by binding EICEs with PU.1[4]. Further, BATF deficiency has a greater influence on the expression of Th9-associated genes than IRF4 deficiency, and the overexpression of BATF can induce IL-9 production in Irf4-/- T cells[15]. Furthermore, BATF- BACH2 complexes promote IL-9 production in Th9 cells[16]. Therefore, IRF4 and BATF co-operatively or independently contribute to Th9 differentiation.
Th17 cells
Th17 cells are induced by IL-6 and TGF-β, express RORγt, produce IL-17, protect the host from bacteria and fungi, and are also related to autoimmunity. The roles of IRF4 and BATF in these cells are well established. Irf4-/- and Batf-/- T cells fail to develop into Th17 cells in vitro. In addition, Irf4-/- and Batf-/- mice are resistant to experimental autoimmune encephalomyelitis, of which the development depends on Th17 cells[4,5]. Both IRF4 and BATF directly bind AICEs around Th17-associated genes such as Il17 , Il21 , Il22 , Il23 r, and Rorc, encoding RORγt. Therefore, IRF4-BATF complexes play fundamental roles in Th17 differentiation and the detailed molecular mechanisms are described as follows.
Treg cells
Treg cells, which are defined based on the expression of Foxp3, produce IL-10 and TGF-β, express CTLA-4, maintain immunological self-tolerance and homeostasis, and protect the host from excessive inflammation, autoimmunity, and allergy. Treg cell-specific Irf4-/- mice show several features of autoimmune disease, such as lymphadenopathy, weight loss, blepharitis, and dermatitis. These mice also exhibit a slight increase in the Treg population in lymphoid tissue. Irf4-/- Treg cells express the same levels of Foxp3 as compared to those in WT Treg cells and possess suppression capacity in vitro, but in these cells, 80% of Treg-associated genes, such as Icos, Il10 , Maf, and Ccr8 , are downregulated. Thus, IRF4 in Treg cells regulates a specific transcriptional program involved in the effector function of these cells[17]. In contrast, BATF deficiency reduces the number of splenic Treg cells. A recent report showed that BATF regulates the development of one Treg cell subtype, specifically CD4+Foxp3+CD44highCCR7low KLRG1+ST2+ Treg cells[18]. However, it is still unclear whether IRF4 and BATF in Treg cells function independently or cooperatively and thus, further studies are needed.
Tfh cells
Tfh cells express C-X-C chemokine receptor type 5 (CXCR5) and programmed cell death 1 (PD-1) , are located in the B cell zone and germinal center of lymphoid tissue, help B cells to undergo affinity maturation through somatic hypermutation, and promote class switch recombination during the germinal center reaction. Tfh cells are induced by IL-6/IL-21-STAT3 signals, are suppressed by IL-2-STAT5 signals, express the master regulator BCL-6, and produce a large amount Tfh cells express C-X-C chemokine receptor type 5 (CXCR5) and programmed cell death 1 (PD-1) , are located in the B cell zone and germinal center of lymphoid tissue, help B cells to undergo affinity maturation through somatic hypermutation, and promote class switch recombination during the germinal center reaction. Tfh cells are induced by IL-6/IL-21-STAT3 signals, are suppressed by IL-2-STAT5 signals, express the master regulator BCL-6, and produce a large amount of IL-21. Interestingly, Irf4-/- mice fail to develop Tfh cells and germinal center B cells. Further, although IRF4 has a great impact on B cell maturation, Irf4-/- T cells are not able to express BCL-6 and IL-21 in a T cellintrinsic manner[4]. As with Irf4-/- mice, Batf-/- mice lack germinal center formation and Tfh cell development capacity. BATF directly regulates the expression of BCL-6 and c-MAF, which are critical TFs for Tfh cell development. However, the reconstitution of BCL-6 and c-MAF in Batf-/- Tfh cells restore Tfh cell functions to support germinal center B cells. Thus, IRF4-BATF complexes function as inducers of BCL-6 and c-MAF during Tfh development[5]
IRF4 and BATF are indispensable for the induction of different genes in diverse Th subsets. Ciofani et al. explored the molecular mechanisms underlying Th17 development using hundreds of next generation sequencing data including ChIP-seq, FRARE-seq, and RNA-seq obtained from WT and TF-deficient Th0, Th1, Th2, and Th17 cells[11]. They identified critical functions of IRF4-BATF complexes with respect to Th17 differentiation and TF networks among IRF4, BATF, STAT3, RORγt, and c-Maf. IRF4-BATF complexes occupied inducible open chromatin that was accessible in Th17 cells but not in naïve CD4 T cells. Further, the FRARE-seq signals on inducible open chromatin were markedly reduced in Irf4-/- or Batf-/- T cells and the binding of STAT3, RORγt, and c-Maf to these regions was almost entirely abolished in Irf4-/- or Batf-/- T cells. Moreover, STAT3 was found to bind part of these open regions and to induce downstream genes including Rorc and Maf. Subsequently, it was demonstrated that RORγt binds Th17-associated genes. In addition, IRF4, BATF, STAT3, and RORγt cooperatively amplify the induction of target genes including themselves (Fig. 2) . c-Maf acts as a transcriptional repressor of the aforementioned four TFs. Importantly, open chromatin develops not only in Th17 cells but also in Th0 cells. Therefore, IRF4-BATF complexes alter the chromatin landscape and facilitate the subsequent recruitment of lineage-specific TFs to the regulatory elements of Th subset-associated genes; thus, IRF4-BATF complexes are able to function as pioneer factors in different Th subsets[11]
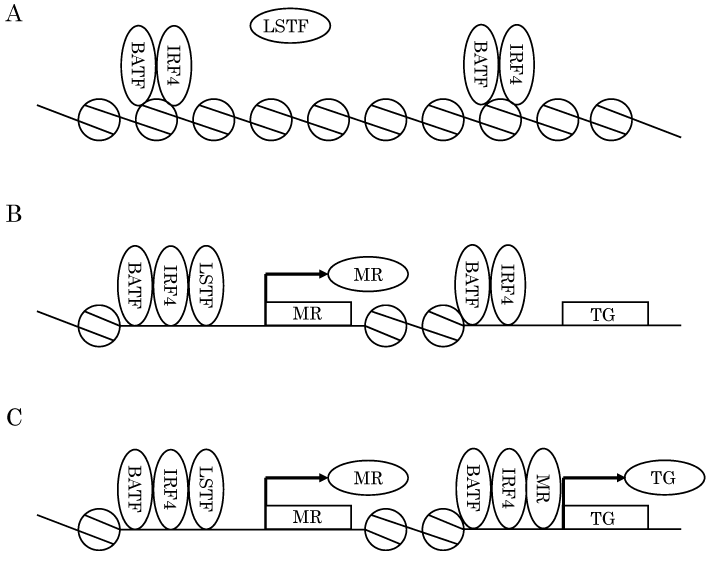
Fig.2 IRF-BATF complexes open the chromatin to initiate a lineage-specific program
(A) IRF4-BATF complexes bind closed chromatin and open the chromatin. (B) Lineage-specific transcription factors (LSTFs) are subsequently recruited to opened chromatin. LSTF and IRF4-BATF complexes cooperatively induce a subset of specific master regulators (MRs) . (C) MR, LSTF, and IRF4-BATF complexes fully activate target gene (TG) loci.
T cells adjust the levels of their clonal expansion and effector cytokine responses according to the level and type of microbial invasion[1]. Excessive expansion and activation cause host damage; therefore, T cells should sense the information about the dose and affinity of the antigen to generate adequate responses and implement a negative regulatory system such as inhibitory molecules like CTLA-4. Thus, TCR signal strength regulates T cell fates including not only their activation but also Th1-Th2, Th17-Treg, and effector T-Tfh balances. WeakTCR signals favor the induction of Th2, Treg, and Tfh cell differentiation, whereas strong signals lead to Th1, Th17, and effector T cell differentiation[3]. TCR signals initially induce kinase cascades in a digital manner; however, the accumulation of downstream signaling cascades including the inducible T cell kinase (ITK) / Ca influx/NFAT pathway and the Atk/mammalian target of rapamycin (mTOR) pathway induce proportional IRF4 expression relative to TCR signal strength[3,19]. The levels of IRF4 in CD8+ T cells directly regulate the graded outcomes, clonal expansion, and effector/ memory fate[19]. In CD4+ T cells, levels of IRF4 and BATF are upregulated by TCR stimulation that reflects the signal strength, as is the case for IRF4 in CD8+ T cells[6]. Recently, how different levels of IRF4-BATF complexes are translated into T cell fates was elucidated. IRF4-BATF-dependent genes show two distinct sensitivity patterns to IRF4-BATF complex levels. Specifically, high sensitivity genes including Gata3 , Prdm1 , and Maf are sensitive to low amounts of IRF4-BATF, whereas low sensitivity genes including Ctla4 , Pparg, and Il12 rb2 require high amounts of IRF4- BATF[6]. In addition, ChIP-seq analysis revealed that all IRF4-BATF-binding sites are not equivalent based on the grade of TCR strength. Low levels of IRF4-BATF complexes preferentially bind high-affinity enhancer elements, which are located around high sensitivity genes; thus, these genes are induced by low numbers of IRF4-BATF complexes. When IRF4-BATF complexes accumulate due to the presence of strong signals, they can bind low-affinity enhancer elements, subsequently inducing low sensitivity genes. Importantly, the affinity of enhancer to IRF4-BATF complexes is defined by the flanking sequence around core AICEs (Fig. 3)[6]. Indeed, the fate choice between effector T cells and Tfh cells in vivo is determined by the affinity of Bcl6 and Prdm1 loci to IRF4[7]. In humans, a single-nucleotide polymorphism (SNP) , rs231735-T, which was reported to be a protective SNP for some autoimmune diseases, is located around CTLA4 gene and comprises a flanking AICE region. This SNP alters the affinity of AICE, from low-affinity to high-affinity, and increases enhancer activity via the sensitivity required for TCR stimulation, which might lead to higher and earlier expression of CTLA-4 under inflammatory conditions compared to that in patients without the SNP; thus, this SNP could protect against autoimmune diseases[6]. Considering that most of human SNPs are located in non-coding regions, further studies on SNPs and alterations in affinity for TFs are needed.
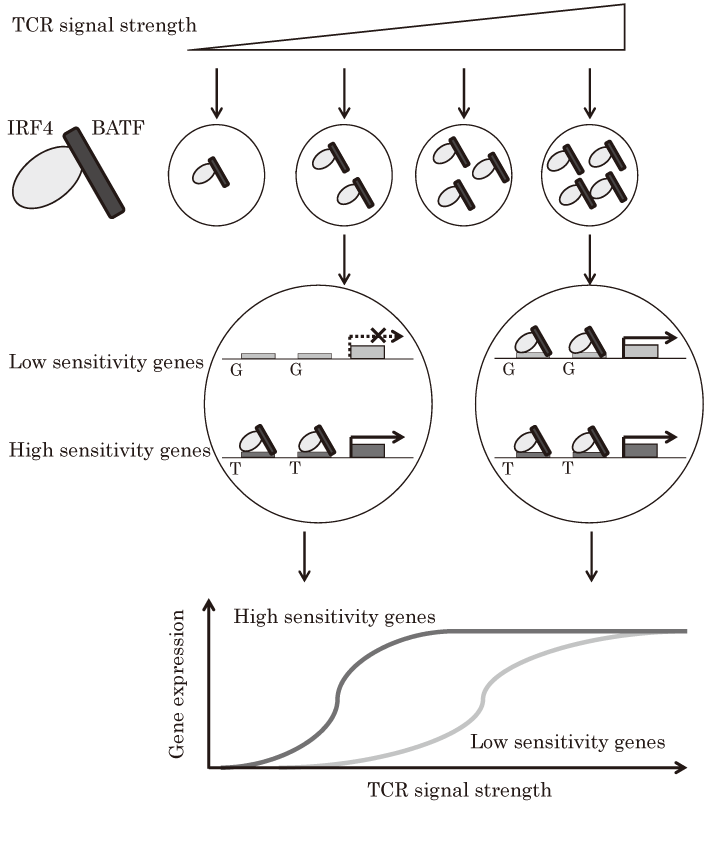
Fig.3 TCR strength translates into diverse gene profiles
Levels of TCR signal strength induce the proportional expression of IRF4-BATF complexes. Upon low-level stimulation, low levels of IRF4-BATF complexes preferentially bind high-affinity sites around high sensitivity genes. Therefore, a fraction of IRF4-BATF-dependent genes is expressed in response to low TCR signal strength. Increased TCR signal strength leads to the accumulation of IRF4-BATF complexes. An abundance of complexes can result in the binding of low-affinity AICE sites. Subsequently, lowsensitivity genes are induced. Therefore, gene expression profiles are regulated by TCR signal strength and the affinity of enhancer sites for IRF4-BATF complexes.
Recent studies have revealed how CD4+ T cell fates are tightly tuned. IRF4-BATF complexes play central roles in T cell fate; however, BATF-BACH2 has also been implicated in this process in the context of IRF4-BATF. BATF-BACH2 complexes function as positive regulators of IL-9 production but also as antagonists of IRF4-BATF complexes on the Il4 promoter. BACH2 is well-known as the central repressor of effector T cell fate; therefore, interactions among IRF4, BATF, and BACH2 should be studied more intensely. Moreover, the pioneer factors involved in Th1 and Treg development are still unknown. Further work on the mechanism underlying T cell fate is thus needed.
AI wrote this manuscript and produced the figures.
I would like to express my gratitude to Prof. Hiroshi Nakajima for critically reading the manuscript. I thank Prof. Kenneth Murphy and Dr. Theresa Murphy for their support and discussion. I also thank Dr. Hiroki Furuya for his constructive comments and feedback.
The author declares no conflicts of interest associated with this manuscript.
Address correspondence to Dr. Arifumi Iwata.
Department of Allergy and Clinical Immunology, Graduate
School of Medicine, Chiba University, 1-8-1, Inohana, Chuouku,
Chiba 260-8670, Japan.
Phone: +81-43-226-2198. Fax: +81-43-226-2199.
E-mail:aiwata@chiba-u.jp
