




Chiba Medical J. 98E:9-18, 2022
doi:10.20776/S03035476-98E-3-P19
〔 Chiba Medical Society Award Review 〕
Hitoshi Shimada1,2)
1) Department of Functional Neurology & Neurosurgery, Center for Integrated Human Brain Science, Brain Research Institute, Niigata University, Niigata 951-8585.
2) Department of Functional Brain Imaging, Institute for Quantum Medical Science, National Institutes for Quantum Science and Technology, Chiba 263-8555.
(Received January 31, 2022, Accepted February 16, 2022, Published June 10, 2022.)
Abnormal proteins deposited in the brain exemplified by amyloid-beta and tau lesions are pathognomonic hallmarks of neurodegenerative disorders. Increasing evidence suggests that these abnormal proteins have pivotal roles in the disruption of the brain environment, leading to the onset of neurodegenerative diseases. The resulting accumulation of abnormal proteins in the brain is considered to represent important target molecules for both diagnostic and therapeutic purposes. In recent years, the development of PET imaging technology for visualizing abnormal brain proteins has been remarkable. Amyloid PET is a most successful imaging technique, and the technology has already matured sufficiently to a practical level to become an indispensable technology in the fields of dementia research and drug discovery. Following amyloid PET, tau PET was developed as a practical pathological imaging technique. Partly because tau lesions are thought to be more closely related to neurological deficits than amyloid-beta deposition, and partly because they are observed in many dementias other than Alzheimer's disease, they are expected to be a game changer in dementia research and drug discovery. In this review, I will focus mainly on tau PET imaging, and especially with PBB3 and PM-PBB3, which were originally developed by our institute, and introduce the current status of their technology development, the characteristics of each tau PET ligand, and the latest findings from clinical PET research. Furthermore, the role and future potential of neuropathological imaging in clinical research and drug discovery for dementia will be discussed.
tau, PET, dementia, PBB3, PM-PBB3
As the population ages, the number of patients with dementia, which is common among the elderly, continues to increase, making it an issue requiring urgent attention. Excluding dementia caused by cerebrovascular diseases, the majority of dementias are thought to be caused by neurodegenerative dementias such as Alzheimer’s disease. The pathological feature of neurodegenerative dementia is the accumulation of various abnormal proteins in the brain. Recently, diseases featuring such abnormal protein accumulations in the brain have been collectively called proteinopathies. Since abnormal proteins in the brain are thought to be deeply involved in the pathogenesis of neurological diseases, they are considered to be important target molecules for diagnosis and treatment.
Attempts to visualize abnormal proteins in the brain have been made since the 1990s, and the most practical method is amyloid imaging using positron emission tomography (PET) and specific ligands. Since the introduction of 11C-Pittsburgh compound-B (PiB) in 2004[1], many 18F-labeled ligands have been developed and have become indispensable technologies for dementia research and drug discovery. On the other hand, tau PET imaging for visualizing the brain accumulation of abnormal tau protein, a component of neurofibrillary tangles that characterizes brain pathology along with amyloid-β in Alzheimer’s disease, has been in practical use since around 2013. With accumulating evidence that tau protein lesions are more closely associated with neurological diseases than amyloid-β, tau imaging is now being used not only in Alzheimer’s disease but also in many non-Alzheimer’s diseases without amyloid-β accumulation. Tau imaging technology has become a standard-bearer for elucidating the pathogenesis of neurodegenerative dementias as well as for driving drug discovery.
In 2013, we reported the development of a firstgeneration tau PET ligand, 11C-PBB3[2]. Since then, we have been conducting clinical PET studies using this ligand to investigate the etiology of neuropsychiatric disorders. In addition, we recently developed 18F-PM- PBB3 (also known as 18F-APN1607), a versatile tau PET ligand with improved drug properties of 11C-PBB3, and reported its preclinical and clinical characteristics [3]. I will introduce the development history and background of 18F-PM-PBB3 as well as the results of clinical trials using 18F-PM-PBB3, and discuss its future prospects.
The development of tau PET ligands has a long history, with a PET ligand called 18F-FDDNP already being developed in the 1990s[4], and clinical studies using this ligand were still being conducted in 2021 [5]. Nonetheless, it has also already been demonstrated that this ligand has low affinity for tau lesions and is not selective for binding to tau lesions because it also shows binding affinity for amyloid-β lesions. Therefore, this ligand is not currently regarded as a tau PET ligand, and caution should be exercised in interpreting reports that feature this PET ligand.
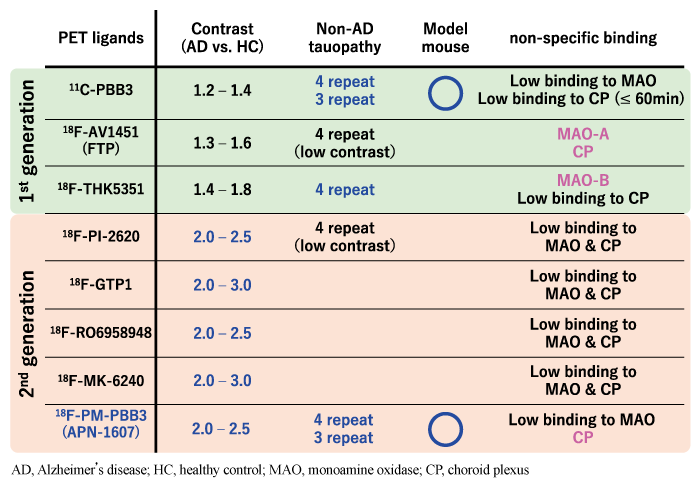
A long time has now passed since the appearance of 18F-FDDNP, and it was not until 2013 that a practical tau PET ligand was developed. The so-called “first generation” tau PET ligands reported during the same period include flortaucipir (also called FTP, AV1451, and T807), which was developed by Siemens and later patented by Eli Lilly and Company[6], and 11C-PBB3 and 18F-THK series developed by Tohoku University (THK-5351, -5117, -5105, -523, etc.) [7]. With the advent of these tau PET ligands, research on the brain pathogenesis of Alzheimer’s disease and other dementias has progressed rapidly. On the other hand, with the progress of tau PET imaging studies, it has also become clear that the first-generation tau PET ligands have various drawbacks that need to be improved. Those early tau PET ligands share the common feature of accumulation around the striatum increasing with age, even in healthy individuals, independent of tau accumulation (off-target binding). These ligands respectively showed a variety of off-target binding sites, including monoamine oxidase (MAO) -A, MAO-B, melanin neurons, choroid plexus, and sagittal sinus, in addition to the striatum[8-10]. For example, in the brains of patients with neurodegenerative diseases, gliosis including reactive astrocytes and activated microglia is observed, and MAO-B is highly expressed in reactive astrocytes. Some first-generation tau PET ligands, such as 18F-THK5351, have binding affinity for MAO-B and may detect gliosis in addition to tau accumulation[10].
Because of these properties of first-generation tau PET ligands, several “second-generation” (or nextgeneration) tau PET ligands have been developed in recent years: Merck’s 18F-MK-6240[11], Piramal’s 18FPI- 2620[12], Roche’s 18F-RO-963[13], Genentech’s 18F-GTP1[14], and Janssen’s 18F-JNJ-64326067[15]. 18F-PM-PBB3 (also known as 18F-APN1607) [3]is a second-generation tau PET ligand developed by the National Institutes for Quantum Science and Technology (QST) of Japan to improve the unfavorable drug properties revealed using the first-generation tau PET ligand 11C-PBB3[2].
In order to understand the development concept of 11C-PBB3, it is necessary to understand the properties of tau protein and the abnormal aggregation of tau protein (tau lesions) observed in various diseases. Tau protein itself is also expressed in the brain of healthy individuals, and it is thought to possess the function of binding to and stabilizing microtubules in neurons. Tau protein is encoded by the microtubule-associated protein tau (MAPT) gene on chromosome 17, and selective splicing produces a total of six isoforms: three 4-repeat tau and three 3-repeat tau. Thus, tau lesions are found in the brains of not only Alzheimer’s disease but also of many other neurodegenerative dementias, and the molecular species that constitute the tau lesions vary depending on the disease (Fig. 1). It has been reported that tau lesions composed of different molecular species have different 3D structures[16]. Therefore, a ligand with high binding affinity for tau lesions in Alzheimer’s disease may not necessarily have good binding affinity for tau lesions in tauopathies other than Alzheimer’s disease. In fact, all tau PET ligands, whether firstor second-generation, have some degree of binding affinity for tau lesions in Alzheimer’s disease and do not interfere with in vivo imaging evaluation; however, binding affinity for tau lesions in non-Alzheimer’s disease tauopathies varies from ligand to ligand. In preclinical and clinical studies, flortaucipir, 18F-MK- 6240, and 18F-RO-948 have all been reported to have low binding affinity for tau lesions in non-Alzheimer’s disease tauopathies[11,12,17,18]. Although there are preclinical and clinical reports claiming the usefulness of 18F-PI-2620 for non-Alzheimer’s disease tauopathies [19,20], there are also reports that the binding affinity of 18F-PI-2620 for tau lesions in progressive supranuclear palsy is lower than that for tau lesions in Alzheimer’s disease, and caution should therefore be exercised in the interpretation of these reports [21].
11C-PBB3 is a tau PET ligand that was developed based on the concept of visualizing various tau lesions in vivo, regardless of the molecular species that make up the tau lesion. Both amyloid-β protein and tau lesions have a higher-order structure called the cross-β-sheet structure, and the amyloid PET ligand is a small-molecule compound with binding affinity to the cross-β-sheet structure of amyloid-β protein. Since amyloid PET ligands cannot visualize tau lesions with the same cross-β-sheet structure, it is assumed that there is some structural difference between tau lesions and amyloid-β protein, which leads to the difference in binding selectivity for both. In order to visualize tau lesions, PET ligands need to show higher binding selectivity for tau lesions than for amyloid-β. Therefore, we first prepared several compounds with different lengths of the basic skeleton as candidate drugs, using the amyloid PET ligand PiB as a lead compound. Next, we evaluated the binding affinity and selectivity of these candidate drugs for tau lesions by directly reacting them with human brain tissue sections. As a result, we found that there is an optimal molecular length for the molecular structure to show high binding affinity and selectivity to tau lesions. We confirmed that PBB3 has the best intracerebral transferability, selectivity to tau lesions, and in vivo stability among the five PBB analogues we prepared, and 11C-PBB3 was then applied to human clinical studies.
In preclinical studies using human brain tissue sections and mouse models, PBB3 was found to have high binding affinity for both Alzheimer’s disease and non-Alzheimer’s disease tauopathies, as well as for tau lesions in a tau-overexpressing mouse model. In subsequent human clinical trials, 11C-PBB3 has been shown to visualize characteristic tau lesions in Alzheimer’s disease and various non-Alzheimer’s disease tauopathies[22-26]. On the other hand, as mentioned above, we also found the following: 1) offtarget binding was observed in the striatum, sagittal sinus, choroid plexus; 2) the detection contrast of tau lesions in non-Alzheimer’s disease tauopathies was lower than that in Alzheimer’s disease; and 3) it is not always easy to determine whether there is an increase in accumulation in individual cases of early-stage elderly or non-Alzheimer’s disease patients. In addition, 11C-PBB3 has been reported as the rare 11C-labeled tau PET ligand. PET ligands labeled with 11C have a short half-life of about 20 minutes, which is convenient for clinical studies because they can be tested on the same day as different PET ligands such as amyloid PET and FDG. On the other hand, PET scan using 11C-labeled ligand can only be performed at facilities that have cyclotrons and PET drug labeling and synthesis technology, and thus the versatility of the test has been a controversial issue.
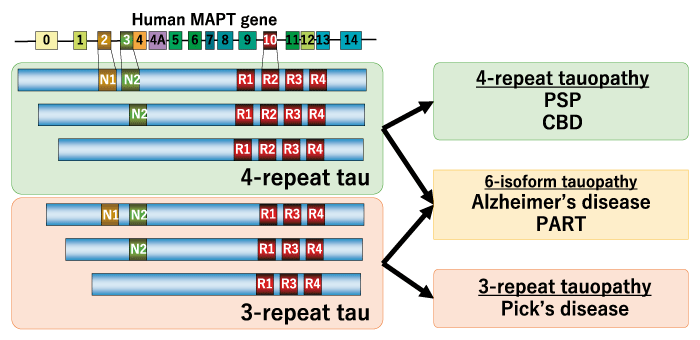
Fig. 1 Molecular species constituting tau lesions in diverse tauopathies. MAPT, microtubule-associated protein tau; PSP, progressive supranuclear palsy; CBD, corticobasal degeneration; PART, primary age-related tauopathy
During the process of human clinical trials using 11C-PBB3, it became clear that there were issues, as described above. In particular, we found that the low detection contrast in non-Alzheimer’s disease may have been due to the low metabolic stability of PBB3 and the fact that a large portion of PBB3 is metabolized even within a few minutes in vivo. The findings from the clinical trials were quickly fed back to basic research in order to improve the metabolic stability of PBB3, which led to the development of 18F-PM-PBB3.
In preclinical evaluation using postmortem brain and mouse models, PM-PBB3 inherited the characteristics of PBB3, the lead compound in development, and showed high selectivity and binding affinity for various tau lesions including Alzheimer’s disease, non-Alzheimer’s disease tauopathies, and mouse models. Based on these results, we decided to use 18F-PM-PBB3 for human clinical research by radiolabeling PM-PBB3 with 18F, which has a longer half-life than 11C, to improve its versatility in clinical practice.
Details of the results of human clinical studies using 18F-PM-PBB3 will be introduced in the next section, but before that, I would like to summarize the characteristics and differences of each ligand (Table 1), which have become difficult to understand due to the emergence of many tau PET ligands. Various first- and second-generation tau PET ligands have been developed; however, the only obvious difference between them is simply the contrast in detection of tau lesions in Alzheimer s disease when compared to healthy subjects. Comparing the ratio of the degree of accumulation of each ligand in the brain of healthy subjects and Alzheimer s disease patients, the secondgeneration ligands have higher ratios than the firstgeneration ligands (about 1.2-1.8 vs. 2.0-3.0), and the difference between the two is easy to discern even at the individual level for the second-generation ligands. In terms of off-target binding to the striatum, MAO-A, MAO-B, etc., most of the second generation ligands, are less obvious; however, some second generation ligands show off-target binding to melanin neurons (18F-MK-6240[11], 18F-PI-2620[12], etc.) and choroid plexus (18F-PM-PBB3[3], etc.). Although all of the recently reported tau PET ligands have sufficient binding affinity for visualizing tau lesions in Alzheimer s disease, few ligands have sufficient binding affinity for non-Alzheimer s disease tauopathies, and only 11C-PBB3 and 18F-PM-PBB3 have been shown to visualize tau lesions in mouse models [2,3].
Table 1 Properties of tau PET ligands
In a human clinical study using 18F-PM-PBB3, pharmacokinetic analysis using arterial blood samples showed that 18F-PM-PBB3 has significantly improved metabolic stability and increased bioavailability in the brain compared to 11C-PBB3. In addition to model analysis such as the 2-tissue compartment model using dynamic imaging data for 150 minutes after drug administration, we also showed that the Standard Uptake Value Ratio (SUVR) using static imaging data for 20 minutes, from 90 to 110 minutes after drug administration, can be used for quantification.
Representative tau PET images with 18F-PM-PBB3 are shown in Figure 2. In healthy subjects with negative amyloid PET, off-target binding to the choroid plexus is observed in some cases, but there is no obvious abnormal accumulation in the brain parenchyma. In the elderly, however, hyperaccumulation reflecting age-related tauopathy may be seen, mainly in the medial temporal lobe, and is pathologically termed Primary Age-related Tauopathy (PART) [27]. In patients with Alzheimer’s disease, a cross-sectional study has shown that in mild and early stage cases, hyper-accumulation is mainly observed in the medial temporal lobe, whereas in advanced cases with severe cognitive impairment, ligand accumulation is spread over a wider range of brain regions and the degree of accumulation is more pronounced (Fig. 3). Although there are differences in the degree of accumulation and non-specific accumulation depending on the ligand, similar accumulation patterns have been reported for all tau PET ligands[7,29,30]. The extent of accumulation of tau PET ligands is very similar to the hypothesized developmental pattern of tau lesions proposed in pathological studies, but some cases do not follow this typical developmental pattern.
Not only in Alzheimer’s disease, but also in non-Alzheimer’s tauopathies, the areas of accumulation expand in disease-specific regions. In Pick’s disease, three-repeat tauopathy, high accumulation of PMPBB3 was found in the inferior frontotemporal lobe. In progressive supranuclear palsy and corticobasal degeneration, four-repeat tauopathies, high accumulation of PM-PBB3 was observed in the brainstem, subthalamic nucleus and striatum, and also in the neocortex in cases with prominent cortical symptoms or in severe cases. The degree of elevated accumulation of PM-PBB3 in disease-specific brain regions correlated well with the clinical severity of cognitive and motor dysfunction. As a result of the significant improvement in detection contrast compared to 11C-PBB3, it was possible to discriminate between healthy subjects and patients with high sensitivity and specificity by assessing the degree of elevated ligand accumulation in individual cases.
Histological examination using biopsy or postmortem autopsy was also performed in some of the cases for which PET imaging was performed. The results showed that in cases pathologically diagnosed as non-Alzheimer’s disease tauopathies including Pick’s disease, progressive supranuclear palsy, and corticobasal degeneration, tau lesions characteristic of each disease were found in brain regions with high accumulation of 18F-PM-PBB3 PET.
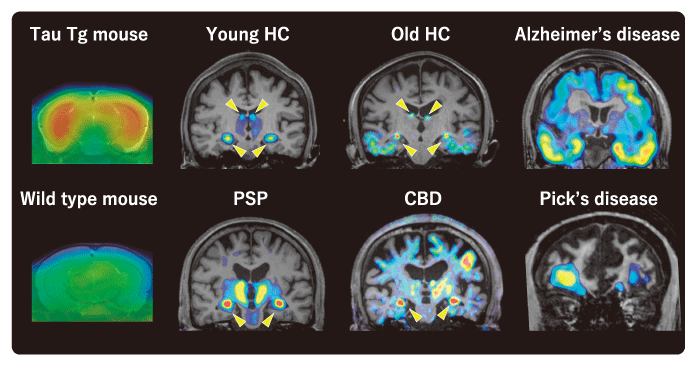
Fig. 2 Representative tau PET image with 18F-PM-PBB3. Arrowheads represent off-target binding to the choroid plexus. HC, healthy control; PSP, progressive supranuclear palsy; CBD, corticobasal degeneration; Tg, transgenic
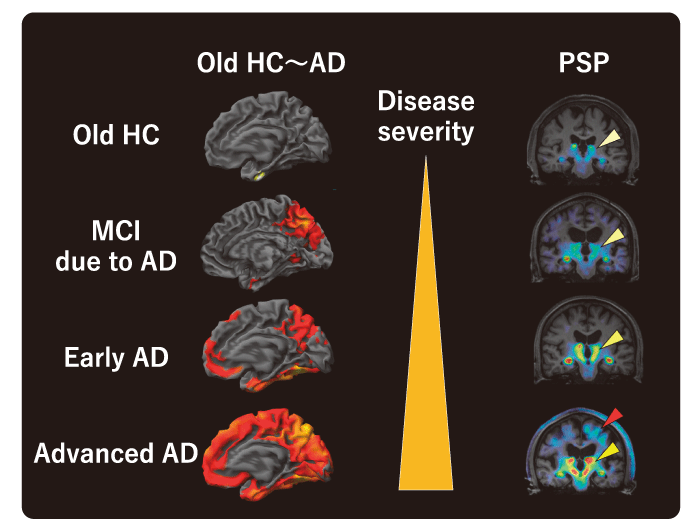
Fig. 3 Association between disease severity and tau PET accumulation. In patients with advanced PSP, remarkable uptake of PM-PBB3 was observed in the cerebral cortex as well as in subcortical regions (red arrowhead). Yellow arrowheads indicate off-target binding to the choroid plexus. HC, healthy control; AD, Alzheimer’s disease; MCI, mild cognitive impairment; PSP, progressive supranuclear palsy
The development history and concept of the novel tau PET imaging ligand 18F-PM-PBB3, as well as its characteristics in preclinical and clinical studies, are reviewed. Tau lesions have been implicated in the pathogenesis of various neuropsychiatric disorders, including neurodegenerative dementia, and drug discovery research has already been conducted targeting tau lesions as a therapeutic target. Looking back at the involvement of amyloid imaging in the development of anti-amyloid-β antibody drugs, it is clear that biomarker technology for early diagnosis, severity assessment, and therapeutic evaluation based on the assessment of brain pathology is required as a fundamental technology for the development of new therapeutic drugs targeting accumulated proteins in the brain. Since 18F-PM-PBB3 has high binding affinity and selectivity for various tau lesions, it is expected to contribute to the differential diagnosis and staging of individual cases. In addition, 18F-PM-PBB3 has a long half-life (approximately 110 minutes) and can be delivered from the factory to hospitals, making it a highly versatile tau PET ligand that can be easily tested at facilities with only PET imaging devices. Clinical studies and clinical trials using 18F-PM-PBB3 are currently being conducted in Japan and overseas, and it is expected that further progress in dementia research will be made and that 18F-PM-PBB3 will be introduced into clinical practice in the near future.
This work was partly supported by Grants-in-Aid for Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS; 15653129 and 19189478), Research and Development Grants for Dementia (16768966 and 19188799) from the Japan Agency for Medical Research and Development, and for Young Scientists (A) (26713031), Scientific Research (C) (18K07543), Scientific Research (B) (16678815), and Scientific Research on Innovative Areas ( “Brain Protein Aging” 26117001) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and for Core Research for Evolutional Science and Technology (CREST; 16810071) from Japan Science and Technology Agency, the Japan Advanced Molecular Imaging Program and the Mochida Memorial Foundation for Medical Pharmaceutical Research, the Life Science Foundation of Japan, and the Kashiwado Memorial Foundation.
The author holds patents on compounds related to the present report (JP 5422782/EP 12884742.3/CA2894994/HK1208672).
Not applicable.
Not applicable.
I am really grateful to all the patients and their caregivers for participation in our studies. I also thank all the staffs including clinical research coordinators, PET and MRI operators, animal care technicians, radiochemists, and research ethics advisers at QST for their assistance with the related projects. In addition, I am thankful to APRINOIA Therapeutics for kindly sharing the precursor of 18F-PM-PBB3. Furthermore, I acknowledge support with the recruitment of patients by Shigeki Hirano at Chiba University, Yuriko Kikkawa at Narita Red Cross Hospital, Kenya Nishioka, Taku Hatano, and Shinji Saiki at Juntendo University School of Medicine, Morinobu Seki at Keio University School of Medicine, Ikuko Aiba at National Hospital Organization Higashinagoya National Hospital, and Yasumasa Kokubo at Mie University. I would also like to express my sincere gratitude to Dr. Satoshi Kuwabara for critically reading this manuscript.
Address correspondence to Dr. Hitoshi Shimada.
Department of Functional Neurology & Neurosurgery,
Center for Integrated Human Brain Science, Brain Research Institute,
Niigata University, 1-757 Asahimachi-Dori, Chuo, Niigata,
Niigata 951-8585, Japan.
Phone: +81-25-227-0683.
Fax: +81-25-227-0822.
E-mail: shimada.hitoshi@bri.niigata-u.ac.jp
