




Chiba Medical J. 90E:37~44,2014
doi:10.20776/S03035476-90E-6-P37
[The Chiba Medical Society Award (2014) ]
Tetsuhiro Chiba
Department of Gastroenterology and Nephrology, Graduate School of Medicine, Chiba University, Chiba 260-8670.
( Accepted September 27, 2014 )
Recent advances in stem cell biology and technologies have enabled the identification of a minor component of tumorigenic cells, termed cancer stem cells or tumor-initiating cells ( TICs ) , in a variety of cancers, including hepatocellular carcinoma ( HCC ) . TICs play a central role in tumor development, metastasis, and recurrence. The analyses of TIC characteristics have revealed the molecular machinery and signaling pathways involved in maintaining them. Although the inhibitors of these molecules and signaling pathways are considered to be promising as TIC-targeting drugs, the establishment of novel therapeutic approaches for the eradication of these cells is yet to be accomplished. In this study, the authors revealed that polycomb group ( PcG ) proteins play an important role in the maintenance of tumor-initiating HCC cells and that short-hairpin RNA and small molecule compounds inhibiting PcG proteins markedly interfere with the tumorigenicity of TICs. Next, we examined whether existing medical drugs could exert their anti-TIC function to promptly apply the research outcomes in clinical settings. Accordingly, we found that disulfiram ( DSF ) , a drug for alcohol dependence, impaired the tumorigenicity of tumor-initiating HCC cells through the activation of the ROS-p38 pathway and the down-regulation of Glypican3 but not through the inhibition of aldehyde dehydrogenase. These results indicate that DSF is a promising therapeutic agent for the eradication of TICs. Based on these findings, a phase I/II clinical trial of DSF was started in patients with advanced HCC.
Hepatocellular carcinoma, cancer stem cell, tumor-initiating cell, polycomb group proteins, ROS-p38 pathway
Hepatocellular carcinoma ( HCC ) represents the sixth most common cancer in the world and is the third most common cause of cancer-related deaths[1]. In Japan, more than 30,000 patients with HCC die every year[2]. Of importance, advanced HCC with extrahepatic spread and/or major vascular invasion has a very poor prognosis. Sorafenib, an oral multikinase inhibitor, blocks tumor cell proliferation by targeting Raf/MEK/ERK signaling and also exerts an antiangiogenic effect by targeting tyrosine kinase receptors, such as vascular endothelial growth factor receptor and platelet derived growth factor receptor[3]. Although sorafenib has been utilized as a standard medical treatment for patients with advanced HCC[4,5], its survival benefit is modest and still unsatisfactory.
Recent advancements in stem cell biology have enabled the identification and characterization of stem cells in a variety of tissues and organs. Moreover, it has been documented that solid tumors such as breast cancer and colon cancer contain a small subset of tumorigenic cells, which can generate new tumors in xenograft transplantation[6,7]. This minor population of cells, termed cancer stem cells ( CSCs ) or tumor-initiating cells ( TICs ) , possesses stem cell-like properties and contributes to a hierarchical structure containing varied progenies in a similar fashion to normal stem cells. Both normal stem cells and TICs share a largely similar surface marker and a molecular machinery controlling self-renewal and differentiation. To explain tumor heterogeneity, two general theories were proposed[8]. The hierarchical model assumes that TICs represent a biologically distinct subset within the total malignant cell population. It is relevant to the designation of malignant tumor-propagating cells as TICs, although it is not necessary that TICs are functionally or genetically homogeneous. According to this model, a pool of TICs can only be maintained by cells that have both TIC potential and the ability to give rise to progeny with a self-limited proliferative capacity. On the other hand, the stochastic model assumes that every cell comprising a tumor has tumor-initiating potential and that their different activities are determined by some stochastically varying intrinsic factor. The recent successful detection and characterization of TICs in a variety of tumors appears to support the hierarchical carcinogenesis theory[9].
Side population ( SP ) cell sorting was developed to enrich hematopoietic stem cells in murine bone marrow [10]. The SP phenotype is determined by the ability to efflux the dye Hoechst 33342 through an adenosine triphosphate ( ATP ) -binding cassette ( ABC ) membrane transporter[11]. This technique has also been widely applied to detect TICs in various cancer cell lines[12]. In our study, only 0.25%-0.80% of HCC cells exhibit the SP phenotype, and these cells show a marked tumorinitiating capacity in xenotransplantation assays using immunodeficient mice[13]. One thousand SP cells were enough to generate tumors in xenotransplantation, while at least 1 × 106 unsorted HCC cells were required for tumor formation, suggesting that TICs are enriched by SP cell sorting by at least a 1000-fold ( Fig. 1A ) . These results indicated that the frequency of HCC SP cells was less than 1%. Therefore, this minority population of SP cells but not non-SP cells might possess tumorigenic potential in these HCC cells.
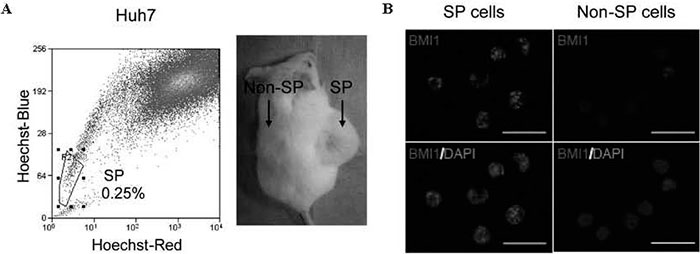
Fig.1 Identification and characterization of SP cells in HCC. (A) Flow cytometry analysis with Hoechst 33342 staining demonstrated that Huh7 cells included 0.25% of SP cells. Only 1 × 103 SP cells purified from Huh7 cells could develop tumors in the NOD/SCID xenograft transplant assay (B) Immunocytochemical analyses of BMI1 expression in SP and non-SP cells purified from Huh7 cells.
More recently, tumor-initiating HCC cells were successfully identified by some cell surface antigens. HCC cells positive for markers, including CD133 [14], CD90[15], CD44[16], epithelial cell adhesion molecule ( EpCAM ) [17], OV6[18], and CD24 [19], were shown to function as TICs in HCC ( Table 1 ) . Of interest, it has been reported that EpCAM and CD90 are independently expressed in primary HCC [20]. The microarray analysis of sorted cells suggested that EpCAM+ cells showed features of epithelial cells, whereas CD90+ cells had features of vascular endothelial cells. HCCs containing EpCAM+ cells are associated with poorly differentiated pathology and a high serum level of alpha-fetoprotein ( AFP ) , whereas the CD90+ HCCs are associated with a high incidence of distant metastasis. Taken together, TICs might possess variable antigenic and functional properties and represent a heterogeneous population of cells.
Table 1 Identification of tumor-initiating HCC cells
Cancer-related molecules and signaling pathways, such as the Polycomb-group ( PcG ) gene products, Wnt signaling, and PI3K/AKT/mTOR pathways, play an important role in the maintenance or augmentation of the self-renewal capability of tumor-initiating HCC cells ( Table 2 ) [21,22]. To develop novel therapeutic approaches for the eradication of TICs, understanding the molecular mechanisms underlying the maintenance of TICs is of importance.
Table 2 Identification of tumor-initiating HCC cells
PcG complexes are the key regulators of epigenetic cellular memory. They establish and maintain cellular identities during embryogenesis, development, and tumorigenesis [23]. PcG complexes can be functionally separated into at least two distinct complexes: a maintenance complex, polycomb repressive complex ( PRC ) 1, and an initiation complex, PRC2. Bmi1, one of the components of PRC1, is essential for maintaining the self-renewal capability of somatic stem cells including hepatic stem/progenitor cells[24]. On the other hand, the overexpression of Bmi1 in hepatic stem/ progenitor cells augments their self-renewal capability and induces tumor development in mice[25]. Of note is that the expression levels of BMI1 in HCC cell lines are well correlated with the proportion of tumor-initiating SP cells ( Fig. 1B ) [26]. Furthermore, the levels of BMI1 expression in HCC are well correlated with the progression and prognosis of the disease[27]. These findings suggest that Bmi1 regulates the self-renewal of both normal stem cells and TICs by repressing the transcription of negative regulator genes for stem cell maintenance, such as Ink4a and Arf[28]
Ezh2, one of the components of PRC2, shows catalytic activity specific for the trimethylation of histone H3 at lysine 27 ( H3K27 ) . Similar to BMI1, EZH2 is also overexpressed in tumor-initiating HCC cells[29]. We previously reported that Ezh2 tightly regulates the self-renewal and differentiation of murine hepatic stem/progenitor cells[30]. In addition, lossof- function assays of EZH2 using short-hairpin RNA and the pharmacological inhibition of EZH2 by an S-adenosylhomocysteine hydrolase inhibitor, 3-deazaneplanocin A ( DZNep ) , showed that both EZH2 -knockdown and DZNep treatment decrease the number of TICs and impair their function ( Fig. 2 ) [31] These findings revealed that tumor-initiating HCC cells are highly dependent on EZH2 for their tumorigenic activity.
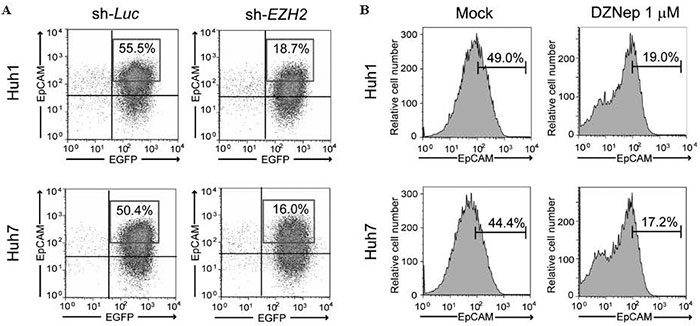
Fig.2 Flow cytometric analyses of EpCAM+ cells.(A) Knockdown of EZH2 significantly decreased the EpCAMhigh fraction in HCC cells.(B) EpCAMhigh fraction was significantly decreased after DZNep treatment in HCC cells.
Although classical anticancer treatment such as chemotherapy and radiation therapy can eliminate proliferating cells in a tumor, TICs often exhibits resistance to these treatments [32,33]. The investigation of treatments targeting TICs has just started and the interference with their self-renewal, survival, migration, or invasion mechanisms are considered a possible strategy[34]. However, no effective therapy targeting TICs has been developed in HCC up to the present. We focused on existing medical drugs showing antitumor effects and examined whether these drugs could eliminate TICs.
Metformin is an oral antidiabetic drug for the treatment of type 2 diabetes mellitus[35]. This drug activates AMP-activated protein kinase ( AMPK ) and thereby contributes to a reduction in hepatic gluconeogenesis and an increase in glucose uptake in skeletal muscles [36]. Of interest, previous studies have suggested that metformin has an anti-cancer effect in various types of malignancies, including breast cancer and ovarian cancer, and even in HCC[37-39]. Various explanations for the anticancer action of metformin have been proposed, such as the activation of AMPK, the inhibition of insulin-like growth factor signaling, and the mTOR pathway[40]. We then examined whether metformin has direct effects on tumor-initiating HCC cells[41]. Sphere formation assays showed that metformin significantly suppressed both the sphere formation ability and the replating activity of tumorinitiating HCC cells in a dose-dependent manner. In addition, metformin treatment reduced the number of EpCAM+ and AFP+ cells in primary spheres ( Fig. 3 ) . This finding indicated that metformin impaired tumorinitiating HCC cells and simultaneously promoted the differentiation toward non-TICs.
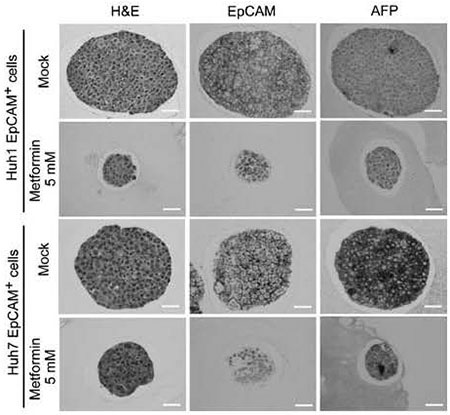
Fig.3 H&E staining and immunocytochemical analysis of spheres derived from EpCAM+ cells. Immunostaining analysis showed that metformin treatment markedly decreased in a number of EpCAM+ and AFP+ cells in spheres.
mTOR signaling also makes a significant contribution to the maintenance of TICs in breast cancer and prostate cancer[42,43]. The aberrant activation of mTOR signaling was also observed in approximately 50% of patients with HCC[44,45]. We demonstrated that metformin treatment apparently inhibited the mTOR pathway by phosphorylating AMPK in tumor-initiating Huh7 cells but not Huh1 cells. This implies that metformin exerts its anti-TIC effect in either an AMPK/ mTOR pathway-dependent or -independent manner. Of interest, metformin was shown to cause cell cycle arrest by downregulating the expression of cyclin D1 and/or upregulating the expression of cyclin-dependent kinase inhibitors such as p21Cip1 without inhibiting the mTOR pathway[46,47]. A recent study revealed a novel mechanism wherein metformin blocks glucagondependent glucose output from hepatocytes by reducing cyclic AMP and protein kinase A levels[48]. Further analyses on the mechanisms of the anti-TIC activity of metformin are required.
Given that the risk of HCC is significantly lower with metformin treatment than with sulphonylureas or insulin in patients with chronic liver disease[49], it is further necessary to examine whether metformin can be used in elimination of TICs in HCC in clinical trials.
For more than 50 years, disulfiram ( DSF ) , an irreversible inhibitor of aldehyde dehydrogenase, has been widely used in alcohol aversion treatmen[t 50]. DSF and its metabolites have been shown to suppress ethanol metabolism mainly through the inhibition of cytosolic aldehyde dehydrogenase 1 ( ALDH1 ) and mitochondrial ALDH2[51]. In addition, recent reports have shown that DSF possesses the potential to target multidrug resistance, angiogenesis, invasion, and proteasome[52]. Of interest, DSF inhibits the tumorigenicity of breast CSCs and enhances the cytotoxicity of anticancer drugs [53]
In this study, we first showed that DSF inhibited the sphere-forming ability of HCC cells and tumor growth in xenograft transplant experiments using non-obese diabetic/severe combined immunodeficient ( NOD/ SCID ) mice in a dose-dependent manner ( Fig. 4 ) . Flow cytometric analysis revealed that the DSF treatment caused a significant decrease in the number of tumor- initiating HCC cells expressing stem cell markers such as CD133 and EpCAM. Given that the knockdown of ALDH1 and ALDH2 had no effect on cell proliferation and sphere-forming ability in HCC cells, it is thought that DSF exerts its anti-HCC function in an ALDHindependent manner.
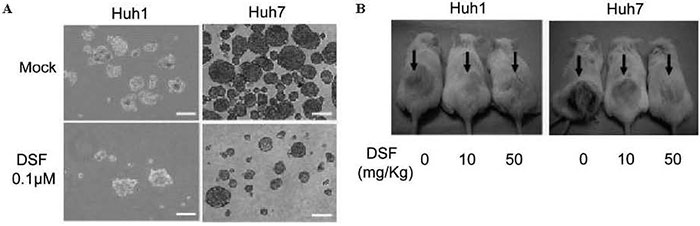
Fig.4 Tumorigenicity of HCC cells treated with DSF in vitro and in a xenograft transplantation model.(A) Sphere-forming ability was significantly impaired in DSF-treated HCC cells.(B) Tumor initiation and growth were apparently suppressed by the DSF treatment.
Although HSCs tightly control intracellular ROS levels to maintain long-term self-renewal and survival, the activation of the p38 MAPK upon an elevation in ROS levels resulted in the exhaustion of HSCs [54,55]. TICs usually exhibit lower intracellular ROS level than corresponding non-TICs[56], and lower ROS levels in TICs result in inferior chemo-sensitivity and radiosensitivity [57]. Previous studies showed that DSF activates the ROS-p38 MAPK pathway and thereby suppresses TIC function[58]. We also confirmed that EpCAM+ HCC cells contained lower ROS levels than EpCAM- cells and that exposure to DSF activated the ROS-p38 MAPK pathway in these cells ( Fig. 5 ) . Of importance, the treatment of EpCAM+ HCC cells with NAC canceled p38 activation. Moreover, SB203580, an inhibitor of the p38 kinase pathway largely restored the tumorigenicity of TICs. These findings indicate that the ROS-p38 MAPK pathway is directly associated with cell growth and the tumor-initiating capability of HCC cells.
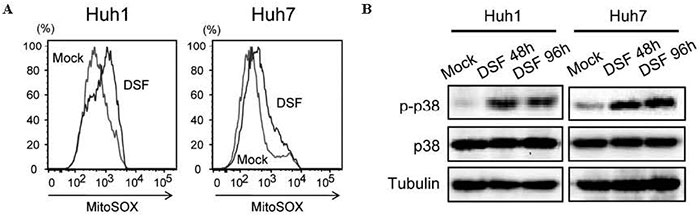
Fig.5 Activation of the ROS-p38 MAPK pathway in HCC cells treated with DSF.(A) Flow cytometric analysis showed an increase in intracellular ROS concentrations after DSF treatment (B) Cells treated with DSF were subjected to Western blot analysis.
Next, we performed microarray analyses using TICs treated with DSF or 5-fluorouracil ( 5-FU ) . The gene set enrichment analysis results support the present biological findings and implicate the activation of p38 in the anti-TIC activity of DSF. Importantly, the 23 genes in the“ liver cancer” category were significantly downregulated after DSF exposure, but none of them were significantly altered after the 5-FU treatment. We focused on one of these genes, GPC3 , because the increased expression of GPC3 was correlated with a poor prognosis among HCC patients[59,60]. The knockdown of GPC3 significantly reduced both the sphere-forming ability and replating activity. Additionally, the immunocytochemical analyses of the large spheres showed a decrease in the number of cells expressing AFP or EpCAM. Taken together, it appears that DSF suppresses the tumorigenicity of tumorinitiating HCC cells, in part, by downregulating GPC3 expression.
Our findings successfully demonstrated that DSF impaired the tumorigenicity of tumor-initiating HCC cells through the activation of the ROS-p38 pathway and through the downregulation of GPC3 ( Fig. 6 ) [61]. DSF might be a promising therapeutic agent for the eradication of tumor-initiating HCC cells. Following the result, we conducted the phase I/II clinical trial of DSF in patients with advanced HCC at Chiba University Hospital after the approval of the institutional review board ( Fig. 7 ) . All patients provided written, informed consent before participation. The study was conducted in accordance with the principles of the Declaration of Helsinki and its amendments in line with Good Clinical Practices. The study was registered in the UMIN Clinical Trial Registry under the number UMIN000008529.
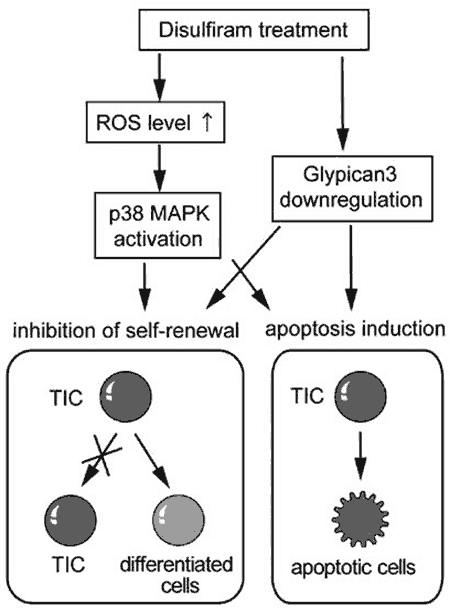
Fig.6 A proposed model for the effect of DSF in targeting tumor-initiating HCC cells.
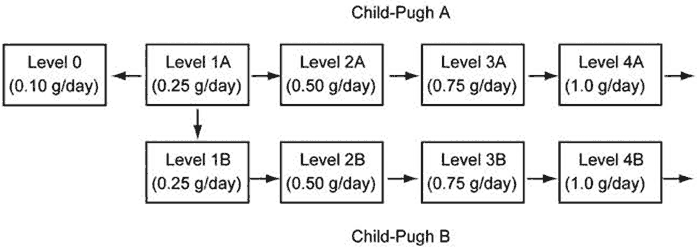
Fig.7 A design for the clinical trial of DSF for patients with advanced HCC.
The CSC theory has given rise to a major paradigm shift in cancer research. It is believed that TICs play a central role in not only cancer development but also metastasis and recurrence after treatment. TIC-specific agents and the combined use of traditional anti-cancer drugs with these targeted TIC-specific agents might offer a promising strategy for the curative treatment of HCC.
I thank Prof. Osamu Yokosuka ( Department of Gastroenterology and Nephrology ) , Prof. Atsushi Iwama (Department of Cellular and Molecular Medicine ) , Prof. Masaru Miyazaki ( Department of General Surgery ) , and the members of HCC group in the Gastroenterology Section of Chiba University Hospital for their valuable discussion and helpful support.
Address correspondence to Dr. Tetsuhiro Chiba.
Department of Gastroenterology and Nephrology, Graduate School of Medicine,
Chiba University,
1-8-1 Inohana Chuouku,Chiba 260-8670, Japan.
Phone: +81-43-222-7171( ext 72010). Fax: +81-43-226-2088.
E-mail: techiba@faculty.chiba-u.jp
