




Chiba Medical J. 93E:17~23,2017
doi:10.20776/S03035476-93E-2-P17
[ The Chiba Medical Society Award(2016) ]
Naoya Mimura
Department of Transfusion Medicine and Cell Therapy, Chiba University Hospital, Chiba 260-8677.
(Accepted January 30, 2017)
Multiple Myeloma(MM) is a malignancy of plasma cells, which remains fatal in spite of recently emerging agents such as proteasome inhibitors and immunomodulatory drugs. The difficulty of the treatment of MM is due to the complex genetic and biological features of this disease and the acquired drug resistance in the context of bone marrow microenvironment. In order to conquer this cancer, we have investigated three promising strategies: targeting the IRE1α-XBP pathway in the unfolded protein response against ER stress, targeting the PI3K/Akt pathway in signal transduction, and targeting histone methyltransferases EZH2 and EZH1 in epigenetic modulation. We propose new combination treatments of novel small molecule inhibitors with proteasome inhibitors which are currently key tools for MM therapy. Based on our data, here we discuss the potential novel therapeutic approaches to achieve the cure of patients with MM.
multiple myeloma, ER stress, signal transduction, epigenetic modulation, proteasome inhibitor
Multiple myeloma(MM) is a plasma cell malignancy characterized by the clonal proliferation of bone marrow (BM) plasma cells, associated with monoclonal protein secreted from tumor cells in the blood and/or urine. Patients with MM suffer from destructive bone lesions, anemia, immunodeficiency, renal failure, hyperviscosity syndrome, and so on. MM accounts for 1% of all cancers and more than 10% of all hematological malignancies in the world. Despite recent advances in treatment including high-dose therapy and novel agents such as bortezomib, thalidomide, and lenalidomide, MM mostly remains incurable due to development of drug resistance in the context of BM microenvironment[1-4]. To overcome this drug resistance, a range of therapeutic approaches has been developed in recent years[5]. Specifically, a new generation of proteasome inhibitors including carfilzomib, ixazomib, and marizomib has been used in the clinic or clinical trials. Pomalidomide, a new class of immunomodulators(IMiDs), has also been added to the clinical options. An HDAC inhibitor panobinostat is currently available in combination with bortezomib and dexamethasone. Moreover, Monoclonal antibodies such as elotuzumab (anti-CS1) and daratumumab(anti-CD38) have been developed for clinical use. However, to achieve the cure of myeloma, different types of strategies are still needed since this disease possesses heterogeneity and complicated biology that enable it to acquire drug resistance. There have emerged novel aspects of therapeutic approaches such as endoplasmic reticulum(ER) stress, signal transduction, epigenetics, and immune response in the BM niche [6]. Among these approaches, we have especially been focusing on ER stress, signal transduction, and epigenetic modulation to conquer this intractable disease.
A cellular organelle endoplasmic reticulum(ER) has many functions to maintain cellular homeostasis, where secretory or membrane proteins are folded properly to form the functional structure with help of molecular chaperones. However, extracellular insults such as low nutrients, hypoxia, and multiple drugs lead to dysfunction of the ER, resulting in the accumulation of misfolded proteins in the ER, thereby triggering ER stress. In reaction to ER stress, cellular response is initiated to reduce the burden of the ER; which is called the unfolded protein response(UPR)[7]. The UPR contains three branches of signaling pathways initiating from three ER transmembrane proteins, i.e. inositol-requiring enzyme 1α(IRE1α), PKR-like ER kinase(PERK), and activating transcription factor 6(ATF6)[8]. In normal conditions, these proteins are associated with molecular chaperone BiP/GRP78 in the ER. In contrast, at the time of ER stress, BiP/GRP78 dissociates from these sensor proteins in order to process the misfolded protein, resulting in induction of the UPR signaling. In the UPR, an endoribonuclease domain of IRE1α is activated to splice the intron with 26 nucleotide from XBP1 mRNA, resulting in a translational frame-shift to turn unspliced XBP1 (XBP1u: inactive form) into spliced XBP1(XBP1s: active form). As a transcription factor, XBP1 regulates genes for protein folding and ER associated degradation (ERAD) to reduce misfolded protein. As a serine/ threonine kinase, PERK phosphorylates eukaryotic translation-initiation factor 2α(eIF2α) in the UPR to inhibit the translation of new protein synthesis, leading to reduction of protein overload in the ER. ATF6 acts as a transcription factor, cleaved into the active form in the UPR. However, under uncompensated stress conditions by these signal transduction, C/EBP homologous protein (CHOP), a pro-apoptotic transcription factor, also known as GADD153, is induced, leading to caspasedependent apoptosis; this is called terminal UPR[9](Figure. 1).
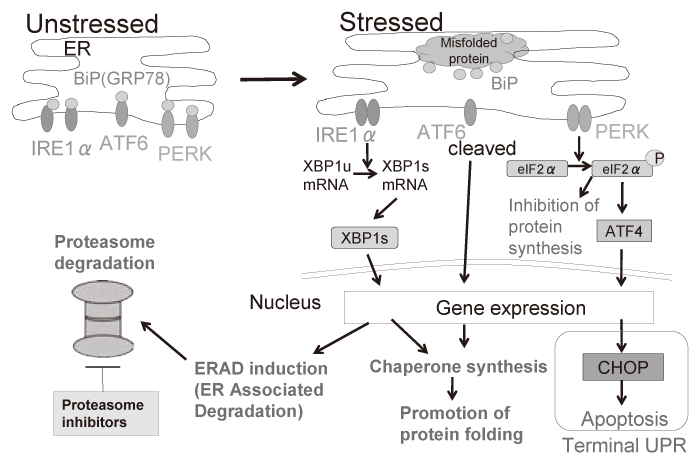
Fig. 1 The overview of the unfolded protein response (UPR) against ER stress
Myeloma cells produce abundant M proteins which trigger high levels of ER stress. This burden of protein synthesis requires strict ER quality control; therefore ER stress and the resulting UPR represent key targets for MM therapy[10,11]. In fact, in clinical practice, the first-used proteasome inhibitor bortezomib induces fatal ER stress in MM cells[12], with great effectiveness in the treatment of MM[13,14]. The ubiquitin-proteasome pathway is responsible for the removal of misfolded proteins accumulated in the ER by ERAD. Bortezomib blocks this pathway, inducing fatal ER stress and apoptosis via upregulated CHOP. Following the success of bortezomib, different classes of proteasome inhibitors such as carfilzomib and ixazomib have been developed for clinical use. Besides the ubiquitin-proteasome pathway, IRE1α-XBP cascade in the UPR has been highly implicated in the pathogenesis of cancers including MM[15]. As for plasma cells, XBP1 is required for the generation of normal plasma cells[16]and is overexpressed in malignant plasma cells[17]. Moreover, knockdown of XBP1 by siRNA sensitizes myeloma cells to stress-induced apoptosis [18]. Additionally, XBP1s overexpression drives MM pathogenesis in murine models[19]. Therefore, targeting XBP1 has been investigated for MM therapy. Particularly, targeting XBP1 splicing by inhibition of IRE1α holds promise as a new therapeutic option which was reported by several preclinical studies including ours [20-22].
We have investigated the preclinical efficacy of a small molecule inhibitor MKC-3946, which blocks XBP1 splicing by inhibition of IRE1α RNase domain (Figure. 2)[20]. Although MKC-3946 induces modest growth inhibition of MM cells in vitro, it induces more significant growth inhibition in vivo. The difference seen between in vivo and in vitro effects is considered to come from more ER stress in vivo due to environmental conditions such as hypoxia or low nutrients. Importantly, MKC-3946 enhances fatal ER stress induced by bortezomib or an HSP90 inhibitor 17-AAG with additive or synergistic effects, since these drugs themselves induce ER stress in MM cells. MKC-3946 also enhances MM growth inhibition by bortezomib in an in vivo xenograft murine model, suggesting clinical application of this combination.
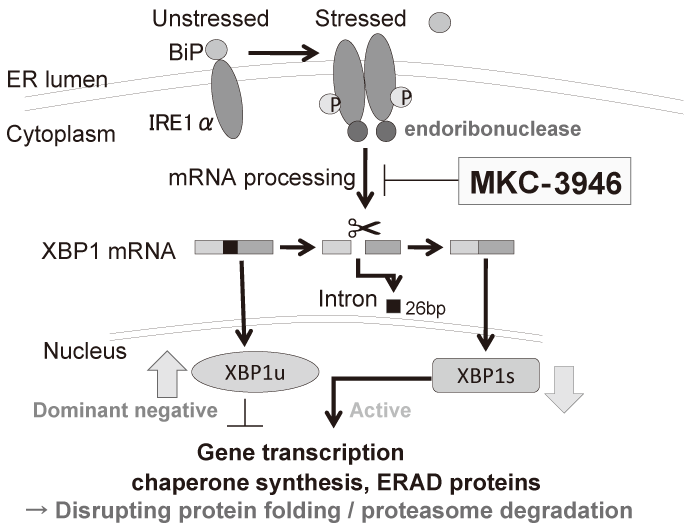
Fig. 2 The mechanisms of action of an IRE1α RNase domain inhibitor MKC-3946
However, it remains controversial whether XBP1 can be a therapeutic target in MM. While low XBP1s level is correlated with the beneficial effects of thalidomide[23], it has been reported that bortezomib is more effective in patients with high XBP1 expresssion [24,25]. In addition, more recently Leung-Hagesteijn et al. have reported that XBP1s is required for bortezomibinduced cytotoxicity in MM, and that XBP1s-negative fraction with arrested secretory maturation becomes resistant to bortezomib[26]. It may be true that MM cells with high-secretory features, which reflect high XBP1s, are more sensitive to proteasome inhibition since these cells have higher burden of protein production with underlining ER stress. However, XBP1 splicing is still considered one of the major adaptive responses of MM cells in order to relieve the acute fatal ER stress induced by proteasome inhibition. Inhibitionof XBP1 splicing is believed to enhance the initial response to bortezomib, especially in MM cells with high XBP1s. However, further investigation is needed to verify its effect in XBP1s-negative population.
Cytokines such as IL-6, IGF-1, VEGF, SDF1, TGFβ, HGF, and TNFα are secreted from BM stromal cells(BMSCs) and MM cells, stimulated by the direct interaction of MM cells with BMSCs. These cytokines vitalize multiple signaling pathways in MM cells, including the phosphatidylinositol 3-kinase(PI3K)/Akt pathway, JAK/STAT3 pathway, RAS/RAF/MEK/ERK pathway, and NF-κB pathways, leading to cell growth, anti-apoptosis, and drug resistance in MM cells [27,28]. Targeting these signal transduction pathways in MM has been tried with many kinds of small molecule inhibitors.
Among them, PI3K/Akt pathway is one of the most crucial signaling in cancers including MM, in terms of cell survival, growth, and drug resistance[29,30]. PI3K includes three classes; class I PI3K, composed of a catalytic subunit p110 and an adaptor/regulatory subunit p85, is activated via upstream receptor tyrosine kinases by various cytokines, phosphorylating and activating the serine-threonine protein kinase Akt. Many substrates such as GSK3α/β and FKHR proteins are phosphorylated by activated Akt, regulating downstream signaling pathways to promote cell proliferation, survival, and anti-apoptosis[31]. In addition, Akt also mediates phosphorylation and activation of mammalian target of rapamycin(mTOR), which plays important roles in cell metabolism and autophagy[32,33](Figure.3). In the pathogenesis of MM, PI3K/Akt pathway plays crucial roles in the context of BM microenvironment. BMSCs stimulates PI3K/Akt signaling cascade in MM cells by the adhesion to MM cells or secreted cytokines such as IL-6 and IGF-1, mediating drug resistance [27,28,34,35]; therefore targeting PI3K/Akt pathway is a promising strategy for the treatment of MM[29,36]. Since Akt is a key player in this pathway, targeting Akt has been investigated in MM. For example, an Akt inhibitor perifosine has preclinical effects on MM cells not only in vitro but also in vivo and enhances cytotoxicity induced by bortezomib[37]. However, perifosine is not selective to Akt signaling, and unfortunately, the clinical trial of perifosine in MM was terminated due to limited clinical response. Therefore, the development of selective Akt inhibitors is still needed.
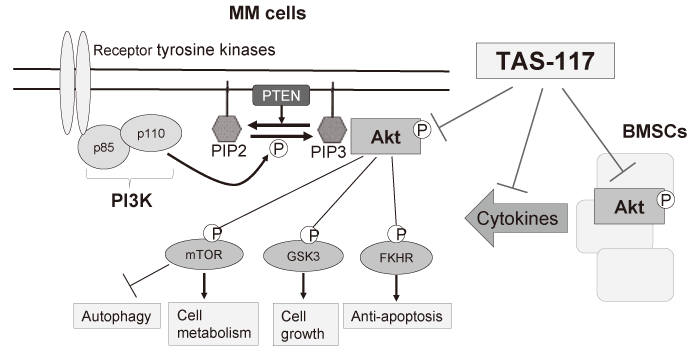
Fig. 3 The schema of the PI3K-Akt pathway and the mechanisms of action of TAS-117
We have shown that a selective and potent allosteric Akt inhibitor TAS-117 induces significant growth inhibition in MM cells both in vitro and in vivo[38] (Figure. 3). Of note, TAS-117 is more potent than perifosine in the BM microenvironment. TAS-117 triggers apoptosis and autophagy, and interestingly ER stress response as well. TAS-117 enhances cytotoxicity induced by proteasome inhibition by bortezomib or carfilzomib, both through inhibition of activated Akt by proteasome inhibitors and through induction of fatal ER stress evidenced by increased CHOP expression. Importantly, synergistic effects between TAS-117 and bortezomib can be observed in an in vivo xenograft mouse model. Moreover, TAS-117 affects MM-initiating cells defined as side population by FACS analysis as well as BM niche cells by inhibiting secretion of IL-6 from BMSCs. Our study proposes the rationale of combination treatment: proteasome inhibition and Akt inhibition in clinical practice.
In the pathogenesis of MM, genetic complexity and molecular heterogeneity are caused by stepwise gene mutations such as chromosomal deletions or gains, translocations, and point mutations. These gene mutations are clearly correlated with clinical features and prognosis in MM[39,40]. Besides,“ epigenetic modifications”, defined as changes in gene expression without alterations in DNA sequence, have been considered crucial in the pathogenesis of MM[41,42]. Many oncogenes and tumor-suppressive genes are regulated by these epigenetic changes; therefore epigenetics is becoming an attractive target for the treatment of cancers. Specifically, epigenetics are categorized into two phenomena: DNA methylation and histone modification.
Histone modification is well investigated and potentially promising in MM. Histones are major protein components of chromatin, where they exist as a complex with DNA to form the nucleosome. The lysine residues of histone tails are modified with various chemical changes such as acetylation, methylation, ubiquitination, phosphorylation, and sumoylation, where gene transcription is altered by these modification[43]. In particular, histone acetyltransferases(HATs) and histone deacetylases(HDACs) determine the acetylation status of proteins and affect physiologic processes involved in cell growth and survival in cancers. Therefore targeting histone acetylation has been clinically applied to develop HDAC inhibitors in the treatment of MM. Panobinostat is now available in clinical practice. Regarding histone methyltransferases, MM set domain(MMSET, also known as WHSC1/NSD2) mainly induces dimethylation of lysine 36 at histone H3(H3K36me2), leading to active gene transcription[44]. It is overexpressed by t (4;14), found in approximately 15% of MM patients and causes pathogenesis of t(4;14) myeloma associated with promoted proliferation of cells[45]. Therefore, MMSET is a promising therapeutic target in t(4;14) myeloma since this population of patients has relatively poor prognosis. However, effective selective inhibitors for MMSET have not yet been developed.
Other histone methyltransferases, enhancer of zeste homolog 2 (EZH2) and its homolog EZH1, can be novel candidates as therapeutic targets in MM. EZH2 and EZH1 are catalytic components of polycomb repressive complex 2(PRC2), which induce H3K27me3 to repress the transcription of target genes and promote tumor growth in some cancers such as lymphoma[46]. EZH2 overexpression in MM has been reported and it correlates with the development from monoclonal gammopathy of undetermined significance (MGUS) to myeloma[47]. Moreover, EZH2 is reportedly indispensable for MM cell growth[48]. Importantly, ubiquitously transcribed tetratricopeptide repeat X chromosome(UTX, also known as KDM6A), a histone demethylase which removes methylation of H3K27, is inactively mutated in a subset of MM samples[49]. These reports suggest that increased H3K27me3 by EZH2 overexpression or UTX inactivation contributes to MM pathogenesis, and that EZH2 and EZH1 act as oncogenes in MM.
We have recently shown that UNC1999, a dual inhibitor of EZH2 and EZH1, demonstrates anti-tumor activities in MM cells both in vitro and in vivo[50]. Furthermore, UNC1999 shows significant synergistic activity with proteasome inhibitors not only in vitro but also in vivo. RNA and ChIP sequencing identifies the target genes of UNC1999 in MM cells including a tumor suppressor NR4A1. The mRNA of NR4A1 is increased and H3K27me3 at the promoter of this gene is suppressed by UNC1999 treatment. NR4A1 is reported to target c-MYC[51]. Overexpression ofNR4A1 inhibits the growth of MM cells associated with suppression of c-MYC, and the combination of UNC1999 and bortezomib further suppresses c-MYC itself and the expression of its target gene sets. These results indicate that the derepression of NR4A1 by UNC1999 and subsequent donwnregulation of c-MYC is one possible mechanism of the cytotoxicity induced by UNC1999 alone and the combination with bortezomib. We have demonstrated that proteasome inhibitors decrease the production of EZH2 mRNA and protein through abrogation of upstream E2F-RB pathway by the blocked degradation of CDK inhibitors p21 and p27 in proteasome; in contrast, proteasome inhibitors maintain the level of EZH1 and global H3K27me3. Being the dual inhibitor of EZH2 and EZH1, UNC1999 works more synergistically than an EZH2 specific inhibitor GSK126 in combination with bortezomib, associated with more significant suppression of H3K27me3. This underlines the importance of dual inhibition of EZH2 and EZH1 in this combination to fully block PRC2 activity(Figure. 4). Our data support the rationale of clinical trials for EZH2 and EZH1 dual inhibitors with proteasome inhibitors [52]
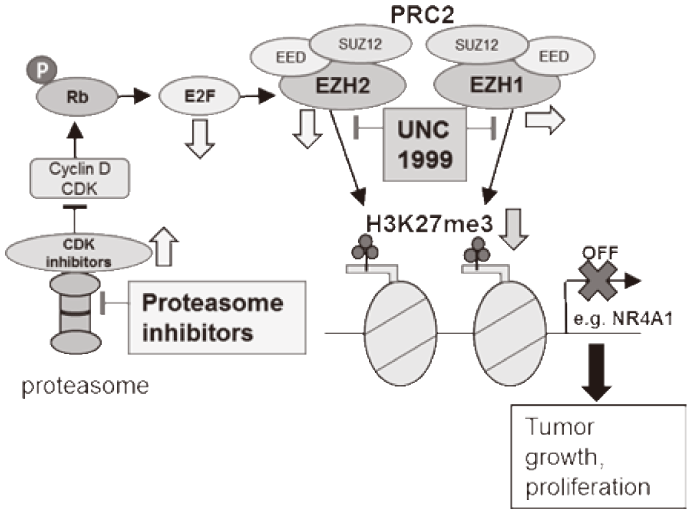
Fig. 4 Synergistic activity of proteasome inhibitors with an EZH2 and EZH1 inhibitor UNC1999
Here we have demonstrated novel therapeutic approaches in MM: targeting the IRE1α-XBP pathway, the PI3K/Akt pathway, and the histone methyltransferases EZH2 and EZH1. These novel treatments are promising especially when combined with proteasome inhibition. Since MM is a heterogeneous disease associated with complex gene abnormalities and multiple signaling aberration, combination treatment is considered very reasonable. Based on our data, clinical trials hopefully will be conducted in the near future. We will continue investigating further treatment options with novel approaches in order to achieve the goal of curing multiple myeloma.
I thank all the investigators for collaborations on these works. I especially appreciate Dr. Ola Rizq for her valuable comments on this review. These works are supported in part by Future Medicine Development at Chiba University, Grants-in-Aid for Scientific Research in Japan, Japan Agency for Medical Research and Development, the Kano-grant from the Japanese society of myeloma, and the International Myeloma Foundation Brian D. Novis Research Junior Grant Award; and grants from Mochida Memorial Foundation, Yasuda Medical Foundation, and Kanae Foundation for the Promotion of Medical Science.
The author has no competing financial interests to declare.
Address correspondence to Dr. Naoya Mimura.
Department of Transfusion Medicine and Cell Therapy, Chiba University Hospital, 1-8-1, Inohana, Chuo-ku, Chiba 260-8677 Japan.
Phone: +81-43-222-7171(ext.71116). Fax: 81-43-226-2478.
E-mail:naoyamimura@chiba-u.jp
