




Chiba Medical J. 102E:19-25, 2026
doi:10.20776/S03035476-102E-1-P19
〔 Chiba Medical Society Award Review 〕
Takayuki Hoshii
Department of Molecular Oncology, Graduate School of Medicine, Chiba University, Chiba 260-8670.
(Received September 16, 2025, Accepted October 16, 2025, Published March 10, 2026.)
Histone methylation is an extensively characterized epigenetic modification tightly linked to transcriptional regulation. In leukemia, the genome-wide distribution and abundance of H3K4 trimethylation (H3K4me3) correlate with the leukemia-initiating cell population, particularly in the KMT2A-rearranged acute myelogenous leukemia (AML) model. KMT2A, a member of the H3K4 methyltransferase family, is a well-established driver of leukemogenesis, and genetic alterations or oncogenic fusions critically contribute to disease onset and progression. Traditionally, the pathogenic functions of these enzymes have been attributed to their catalytic activity. However, accumulating evidence indicates that KMT2A and other H3K4me3 methyltransferases promote leukemia progression through non-catalytic mechanisms. These include scaffolding functions that organize transcriptional complexes and regulate cellular metabolism independent of histone modification. This review summarizes the current knowledge on the non-catalytic functions of H3K4 methyltransferases, with a focus on KMT2A and related proteins, SETD1A/B, and discusses how targeting these mechanisms may provide novel opportunities for leukemia therapy.
H3K4, Methyltransferase, Leukemia, Catalytic, Non-catalytic
Histone lysine methylation occurs in mono-, di-, and trimethylated states, each linked to distinct chromatin landscapes and gene regulatory functions in eukaryotes. Histone H3 lysine 4 (H3K4) is one of the most well-studied epigenetic modifications. H3K4me1 is enriched in enhancers, H3K4me2 spans both promoters and enhancers, and H3K4me3 is concentrated at the transcription start sites of actively transcribed genes [1] . Although these modifications play essential roles in normal development and lineage specification, their aberrant regulation is increasingly recognized as a hallmark of human disease.
Aberrant patterns of H3K4 methylation are frequently observed in cancers, where they contribute to transcriptional programs that sustain uncontrolled proliferation, impaired differentiation, and resistance to therapy. Alterations in H3K4 methylation are particularly prominent in hematological malignancies. For instance, leukemia stem and progenitor cells in acute myelogenous leukemia (AML) model often display distinct H3K4me3 landscapes that correlate with their self-renewal potential [2] . Moreover, atypical distributions such as broad H3K4me3 domains or “bivalent” promoter states can enforce abnormal transcriptional priming, thus providing a permissive environment for leukemic transformation from normal hematopoiesis [3-6].
Enzymes that catalyze H3K4 methylation are central to these processes. In mammals, the KMT2 family of proteins, including KMT2A and SETD1A/ B, is responsible for establishing different methylation states. KMT2A rearrangements (KMT2A-r), including fusion with other genes or partial tandem duplications, are defining drivers of acute leukemia, and SETD1A/ B has been implicated in leukemic cell survival [7-9] . Importantly, the contribution of these enzymes extends beyond their enzymatic activity on histones and involves scaffolding and regulatory functions that play decisive roles in tumorigenesis.
This duality of catalytic and non-catalytic functions highlights the complexity of H3K4 methylation in leukemia biology and motivates therapeutic exploration beyond the inhibition of catalytic activity alone.
KMT2A (MLL1), a mammalian homolog of the Drosophila trithorax protein, has emerged as a central regulator of developmental gene expression and a critical driver of leukemogenesis. Its canonical role involves the regulation of clustered homeobox (Hox) genes, which are indispensable for anterior-posterior patterning and vertebrate organogenesis [10] . At the molecular level, KMT2A functions within a large multiprotein complex containing WRAD (WDR5,RBBP5, ASH2L, and DPY30) and cofactors such as Menin (MEN1) and LEDGF (Fig. 1A) [11-13] . Through its C-terminal SET domain, KMT2A catalyzes H3K4 methylation, thereby maintaining transcriptionally competent chromatin states.
Despite the importance of its catalytic activity, genetic studies have demonstrated that its enzymatic function alone does not fully account for the essentiality of KMT2A. Knock-in mice engineered to lack the SET domain are viable, whereas complete Kmt2 a knockout results in embryonic lethality [10,14] . These findings underscore the indispensability of KMT2A’s non-catalytic functions during development. Mechanistically, these non-catalytic roles are mediated by the N-terminal region, which contains DNA/chromatin interaction motifs and a MEN1-binding motif (MBM) (Fig. 1A) [15] . This N-terminal module orchestrates the recruitment of KMT2A to target loci, stabilizes chromatin occupancy, and facilitates gene regulation independent of enzymatic activity.
The relevance of these non-catalytic mechanisms is particularly evident in the pathogenesis of KMT2A-r leukemia. Chromosomal translocations involving KMT2A generate oncogenic fusion proteins that invariably lack the C-terminal SET domain, but preserve the N-terminal MBM. These fusion proteins aberrantly recruit transcriptional elongation machinery, including the Super Elongation Complex (SEC), thereby driving sustained expression of leukemogenic programs such as the HOXA cluster and MEIS1 (Fig. 1B) [13] . Thus, the module essential for normal development was co-opted for malignancy, providing a unifying principle linking developmental biology with oncogenesis.
The therapeutic implications of these mechanistic insights are significant. Traditional approaches targeting the SET domain are ineffective against KMT2A-r leukemia because fusion proteins lack this enzymatic module. Instead, pharmacological disruption of the MEN1-KMT2A interaction has emerged as a rational strategy [16] . MEN1 inhibitors abrogate the recruitment of KMT2A fusion proteins to the chromatin, suppress leukemic transcription, and induce differentiation [17] . Revumenib, the first-in-class MEN1 inhibitor, received FDA approval in 2024 for the treatment of relapsed/ refractory KMT2A-r leukemia, marking a milestone in epigenetic therapy [18] . Ongoing studies are exploring the mechanisms of resistance such as secondary mutations in MEN1 or KMT2A, adaptive rewiring of chromatin complexes, and lineage plasticity, which will likely inform the development of next-generation inhibitors or combination regimens.
In summary, KMT2A is a chromatin regulator whose developmental and oncogenic functions are critically shaped by both catalytic and non-catalytic modalities. The paradigm shift from enzymatic inhibition to targeting protein-protein interactions highlights the importance of understanding non-canonical mechanisms in epigenetic therapy. Continued mechanistic dissection of KMT2A’s multifaceted functions will further refine therapeutic strategies and expand the conceptual framework of chromatin-targeted oncology.
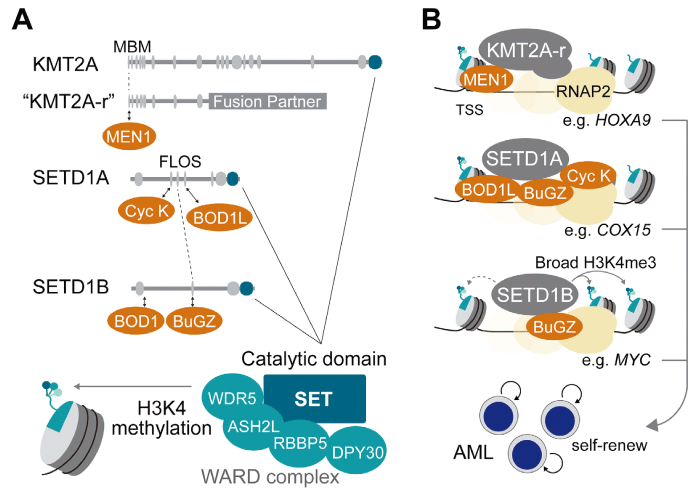
Fig. 1 Roles of KMT2A-r, SETD1A, and SETD1B in AML. A)
Partner proteins (orange) involved in non-catalytic functionsand their interacting domains. The catalytic SET domain and the WARD complex responsible for H3K4 methylation are also indicated. B) Illustration of representative H3K4 methyltransferases in transcriptional activation in AML cells. Such transcriptional activations promote the self-renewal of AML-initiating cells.
While KMT2A-fusion has served as a paradigm for understanding how the non-catalytic function of chromatin regulators can be hijacked in leukemogenesis, SETD1A represents another compelling example of this principle with direct therapeutic implications (Fig. 1A-B) . SETD1A (KMT2F) is also a member of the KMT2 family, which mediates H3K4me3 through its SET domain, in concert with WRAD cofactors. However, studies on AML, particularly in the context of KMT2A-r, revealed that the oncogenic role of SETD1A is largely independent of its catalytic activity.
Instead, SETD1A exerts critical non-enzymatic functions via its central FLOS (Functional location on SETD1A) domain, which interacts with the Cyclin K/ CDK12 complex to promote RNA polymerase II pause release and transcriptional elongation of genes involved in DNA damage repair and mitochondrial metabolism (Fig. 1A, B) [8,19] . This mechanism is indispensable for the survival of KMT2A-r AML and other cancer cells, and its disruption leads to impaired DNA repair, metabolic stress, cell cycle control, and loss of cell viability [8,20,21] .
In addition to Cyclin K/CDK12, BuGZ and BUB3 have been identified as FLOS domain-interacting partners [22] . BuGZ contains an intrinsically disordered region that mediates interactions with BUB3 and SETD1A. Because BuGZ and BUB3 are distributed in both enhancers and promoters, their association with SETD1A may further potentiate transcriptional activation.
BOD1L is another critical partner of the FLOS domain (Fig. 1A) [23] . BOD1L is highly expressed in AML and recruits SETD1A to chromatin. Notably, BOD1L depletion recapitulated the changes in gene expression observed in SETD1A knockout cells, establishing BOD1L as a potential therapeutic target in SETD1A-dependent cancers. Importantly, these effects occurred without alterations in H3K4 methylation, highlighting a unique therapeutic vulnerability distinct from canonical enzymatic inhibition. In contrast, BOD1L is required for SETD1A-dependent H3K4 methylation during the recruitment of RIF1 to double-strand breaks in other cell types [24] . Taken together, BOD1L is an essential component of SETD1A with both catalytic and non-catalytic activities.
From a clinical standpoint, these findings suggest that targeting SETD1A should focus not on its SET domain but on its protein-protein interaction interfaces. In particular, pharmacological disruption of the SETD1A-Cyclin K/CDK12 axis may recapitulate the anti-leukemic effects observed upon genetic depletion. Given that Cyclin K/CDK12 is a transcription-associated kinase required for the maintenance of genomic stability, inhibitors that perturb this pathway may synergize with DNA-damaging agents, such as topoisomerase inhibitors or PARP inhibitors, thereby enhancing therapeutic efficacy [25] . Moreover, SETD1A-driven programs that sustain mitochondrial function and glutamate metabolism highlight the possibility of combining SETD1A-targeted strategies with metabolic interventions.
Unlike KMT2A, where MEN1 inhibitors have already entered the clinic and established a proof-of-concept for targeting non-catalytic mechanisms, SETD1A-directed therapies remain in the preclinical stage. As drug discovery efforts increasingly focus on targeting protein complexes and interaction domains, SETD1A has emerged as a high-priority candidate for the development of next-generation leukemia therapies.
Taken together, SETD1A illustrates a broader paradigm in cancer biology; non-catalytic chromatin regulatory functions can be exploited as therapeutic targets. Building on the clinical success of MEN1 inhibitors in KMT2A-rearranged leukemia, translational research aimed at disrupting SETD1A’s FLOS domain-mediated functions may define the next frontier in precision epigenetic Unlike KMT2A, where MEN1 inhibitors.
SETD1B (KMT2G), a paralog of SETD1A, forms a human H3K4 methyltransferase complex that is primarily responsible for H3K4me3 (Fig. 1A) . Although SETD1A is broadly required for cancer cell proliferation, it is rarely mutated. SETD1B is frequently altered in human malignancies, most notably in B-cell lymphoma, highlighting its disease relevance through distinct molecular mechanisms [26] .
Emerging evidence indicates that SETD1B has both canonical and non-canonical functions in tumorigenesis. Canonically, SETD1B regulates broad H3K4me3 domains essential for the transcriptional control of robustly expressed genes. Genetic studies have demonstrated that the catalytic activity of SETD1B is indispensable for neuronal development, spermatogenesis, and oogenesis, underscoring its non-redundant role in normal physiology [27-29] . In hematopoietic malignancies, our group showed that SETD1B-dependent H3K4 methylation supports cytokine-independent growth and maintains MYC expression, establishing its catalytic activity as a promoter of leukemogenesis (Fig. 1B) [9] . Importantly, this function is mechanistically distinct from that of SETD1A, whose enzymatic activity is dispensable in AML, suggesting differential reliance on catalytic versus scaffolding functions between the two paralogs.
In parallel, non-canonical functions of SETD1B have emerged. Unlike SETD1A, which interacts with BOD1L and Cyclin K to coordinate transcription elongation, SETD1B specifically associates with BOD1, a cytoplasmic protein involved in breast cancer progression (Fig. 1A) [30] . The loss of either SETD1B or BOD1 upregulates fatty acid metabolism-related genes without affecting global H3K4me3 levels, suggesting that this pathway relies on a non-catalytic, context-specific mechanism. In contrast, BOD1 is dispensable in AML, consistent with the catalytic function of SETD1B in this setting [9] . These findings support the concept that SETD1B contributes to tumor biology via dual modalities, catalytic and non-catalytic, depending on the cellular context and interacting partners.
In embryonic stem cells, SETD1A and SETD1B are functionally redundant and establish broad H3K4me3 peaks [31] . However, in leukemia models, the deletion of the SET domain in SETD1A combined with SETD1B depletion does not yield additive effects [9] . This divergence suggests context-dependent utilization of SETD1A and SETD1B, with SETD1B’s enzymatic role being more universally required across cell types, whereas SETD1A exerts highly cell type-specific enzymatic functions.
In B-cell lymphoma, recurrent SETD1B mutations have been shown to impair the expression of proapoptotic genes through their catalytic functions, thereby conferring resistance to apoptosis [26] . Notably, the same study suggested that inhibition of the histone demethylase KDM5 could restore sensitivity to venetoclax, highlighting the therapeutic potential of rebalancing H3K4 methylation dynamics in this disease context.
In contrast, our studies on AML with FLT3 mutations and RAS activation demonstrate that SETD1B’s catalytic activity is indispensable for maintaining MYC expression and promoting leukemic proliferation [9] . Interestingly, inhibition of KDM5C can partly restore both cell proliferation and H3K4 methylation in SETD1B-deficient AML cells. Thus, although SETD1B mutations exploit its methyltransferase function to suppress apoptotic pathways in B-cell lymphoma, the same enzymatic activity drives oncogenic transcriptional programs in AML.
From a translational perspective, these findings highlight the need for disease-specific therapeutics. In AML, targeting the SETD1B SET domain with inhibitors or degraders can directly disrupt MYC-driven leukemogenesis. However, in B-cell lymphoma, therapies aimed at overcoming resistance may be more effective when focused on epigenetic modulators such as KDM5 inhibitors, which can synergize with venetoclax by counteracting the apoptosis-resistant state induced by mutant SETD1B.
Collectively, these observations highlighted the mechanistic versatility of SETD1B and its context-dependent oncogenic functions. Defining whether SETD1B mutations act through catalytic or non-catalytic pathways in a given malignancy is critical for tailoring therapeutic strategies and optimizing clinical outcomes.
H3K4 methylation modifiers indicate how chroma-tin regulators contribute to leukemogenesis through both catalytic and non-catalytic mechanisms. Insights from studies on KMT2A, SETD1A, and SETD1B highlight that oncogenic functions are highly context-dependent and cannot be explained by enzymatic activity alone.
These findings emphasize the need for therapeutic approaches that extend beyond conventional catalytic inhibition to include the disruption of critical interaction networks or rebalancing of chromatin states. Defining the precise modality-enzymatic or scaffolding-through which each modifier drives malignancy is essential for designing disease-specific interventions and advancing precision epigenetic therapies.
T.H. was supported by JSPS KAKENHI grant JP22H03099, the Takeda Science Foundation, and AMED P-PROMOTE grant JP25ama221144.
The author declares no conflict of interests.
Not applicable.
Not applicable.
I thank Dr. Kaneda for critically reading this manuscript.
Address correspondence to Dr. Takayuki Hoshii.
Department of Molecular Oncology, Graduate School of
Medicine, Chiba University, 1-8-1, Inohana, Chuou-ku, Chiba
260-8670, Japan.
Phone/Fax: +81-43-226-2039.
E-mail: hoshiit@chiba-u.jp
