




Chiba Medical J. 102E:27-34, 2026
doi:10.20776/S03035476-102E-1-P27
〔 Chiba Medical Society Award Review 〕
Chiaki Iwamura1,2)
1) Department of Immunology, Graduate School of Medicine, Chiba University, Chiba 260-8670.
2) Synergy Institute for Futuristic Mucosal Vaccine Research and Development, Chiba University, Chiba 260-8670.
(Received October 2, 2025, Accepted October 16, 2025, Published March 10, 2026.)
Although the immune system is inherently designed to eliminate microbes, its development depends on its presence. The overall aim of this review is to highlight how host-microbe interactions, from commensal-derived signals to viral infections, shape immunity and influence both protective and pathological immune responses. Commensal microorganisms and their metabolic products have been shown to be indispensable for the maturation of the immune system. We demonstrated that peptidoglycan, a component of the Gram-negative bacterial cell wall, circulates in the blood of mice under steady-state conditions and is essential for maintaining normal hematopoiesis through the activation of the pattern recognition receptor NOD1 expressed in bone marrow mesenchymal stem cells. Moreover, NOD1 is expressed in peripheral lymphocytes, where it regulates their proliferation and survival by controlling the function of STAT5. The NOD-STAT5 axis is required to protect the host from protozoan infections. Therefore, NOD1 contributes to host protection by shaping the adaptive immune system. In addition to immunological homeostasis, understanding the immune responses to invading pathogens is important. During the COVID-19 pandemic, we showed that SARS-CoV-2 infects pulmonary endothelial cells and that non-classical macrophages recruit and produce thrombospondin-1, which promotes thrombosis, leading to impaired oxygenation and systemic deterioration. These lung thrombi contained platelet factor MYL9/12, and the serum level of MYL9 correlated with disease severity. Furthermore, we found that a subset of severe cases harbored autoantibodies against type I interferons, resulting in defective antiviral responses and poor viral control. These findings not only provide insight into the mechanisms of COVID-19 severity but also offer important perspectives against future viral pandemics.
NOD1, hematopoiesis, COVID-19, MYL9, Anti-type I IFN antibody
Although the immune system has been developed as a defense mechanism against invading microbes, microbial presence is indispensable for the maturation and maintenance of immune homeostasis[1]. Immune cells express pattern recognition receptors (PRRs) that detect microbial components and induce appropriate immune responses[2]. Commensal microorganisms generate metabolites that influence the development and function of immune cells, particularly in mucosal tissues such as the intestine[3]. However, how hematopoietic stem and progenitor cells (HSPCs) located deep within the bone marrow and distant from microbial niches perceive microbial products under steady-state conditions remains an unresolved question. (I) Our group addressed this question and demonstrated that bacterial cell wall components, particularly peptidoglycan, circulate in the blood and influence hematopoiesis via the PRR “NOD1” [4]. (II) We further showed that NOD1 not only responds to microbial ligands but also recognizes endogenous ligands, thereby exerting microbe-independent functions in adaptive immune regulation[5]. These findings suggested that NOD1 is a critical regulator of immune homeostasis.
The immune system is essential for defense against infections; however, its excessive responses can sometimes exacerbate disease symptoms. During the COVID-19 pandemic, understanding the immunopathology of infectious diseases has become an urgent priority. (III) We revealed that a subset of monocytes recruited to blood vessels damaged by SARS-CoV-2 infection contributed to disease severity by promoting thrombosis. In addition, we demonstrated that the serum level of platelet factor MYL9 serves as a biomarker for evaluating COVID-19 severity [6]. (IV) Furthermore, autoantibodies against type I interferons impair antiviral responses, resulting in severe disease[7]. In this review, we summarize these advances, focusing on our recent work on NOD1-mediated immune regulation and the mechanisms of severe COVID-19.
Germ-free (GF) mouse studies have highlighted the essential role of commensal bacteria in regulating peripheral immune responses; however, their influence on bone marrow (BM) hematopoiesis remains unclear. Comparative analyses of HSPCs between GF and specific-pathogen-free (SPF) mice revealed that the absence of microbiota leads to reduced numbers of hematopoietic stem cells (HSCs), common myeloid progenitors (CMPs) , and common lymphoid progenitors (CLPs) [4]. These findings suggest that microbial signals influence the broader HSPC pool in BM.
Focusing on microbial pattern recognition receptors, we examined the role of nucleotide-binding oligomerization domain (NOD) proteins. NOD1 and NOD2 detect distinct bacterial peptidoglycan motifs, meso-diaminopimelic acid and muramyl dipeptide, respectively[8]. Serum from GF mice contained markedly lower concentrations of both NOD1 and NOD2 ligands than SPF controls. Importantly, HSPCs expressed significant levels of NOD1 mRNA, but not NOD2, prompting the evaluation of NOD1 ligand effects in vivo. The oral administration of an NOD1 ligand to GF mice restored HSC, CMP, and CLP numbers to SPF-equivalent levels, implicating NOD1 signaling as a positive regulator of hematopoietic progenitor homeostasis.
Direct stimulation assays indicated that NOD1 ligand alone did not drive HSPC proliferation or differentiation in vitro, suggesting an indirect mechanism. A further investigation focused on bone marrow mesenchymal stromal cells (MSCs) , which are critical niche components for HSC regulation. MSCs selectively expressed NOD1, and its expression increased upon ligand stimulation. Furthermore, NOD1 activation upregulated the transcripts of hematopoietic cytokines, including IL-3, IL-7, Flt3L, stem cell factor (SCF) , and thrombopoietin (ThPO). In vivo, oral delivery of NOD1 ligand to GF mice restored serum cytokine levels (IL-7, Flt3L, SCF, and ThPO) to those observed in SPF animals. Thus, NOD1-mediated signaling in stromal cells drives systemic cytokine induction, indirectly supporting HSPC expansion and steady-state hematopoiesis.
Collectively, these findings demonstrated that commensal bacteria-derived NOD1 ligands act through BM stromal niches to promote hematopoietic cytokine production and sustain the HSPC pool (Fig. 1, Upper panels). This work reveals a critical pathway by which the microbiota contributes to hematopoietic regulation in the BM, expanding our understanding of host-microbe interactions and highlighting NOD1 as a key mediator linking commensal bacteria to hematopoietic homeostasis.
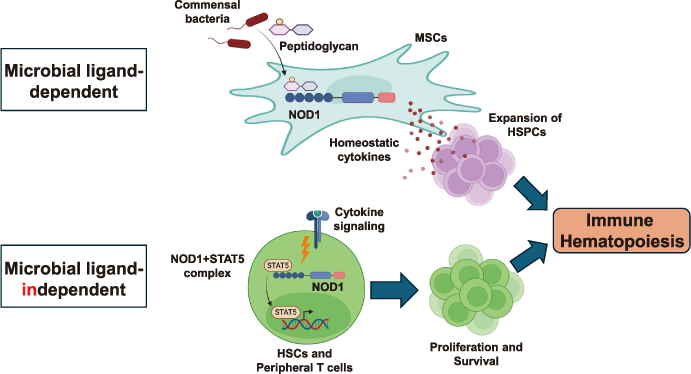
Fig. 1 Microbial ligand-dependent and independent regulation of immune homeostasis.
In bone marrow mesenchymal stem cells (MSCs), NOD1 senses the peptides glycan derived from commensal bacteria. Upon ligand recognition, MSCs produce hematopoietic cytokines that promote the proliferation of hematopoietic stem cells and progenitor cells (HSPCs) (upper panel) . In addition, cytokine stimulation induces cytosolic interactions between NOD1 and STAT5 in HSCs and peripheral T cells, whose binding is essential for their proliferation and survival (lower). These pathways ensure optimal hematopoiesis.
Investigations into the role of NOD1 in hematopoiesis extended to NOD1-deficient (NOD1-/-) mice, which revealed selective defects in lymphoid but not myeloid compartments[5]. While overall BM cellularity remained comparable to that of wild-type (WT) controls, NOD1-/- animals exhibited reduced thymic, splenic, and lymph node cellularity. A phenotypic analysis revealed broad reductions in T, B, and NK cells, whereas dendritic cells, macrophages, and granulocytes were minimally affected. Within the BM, NOD1-/- mice had diminished HSCs and CLPs, but normal CMPs. These data highlighted a specific requirement of NOD1 in steady-state lymphopoiesis.
BM transfer experiments clarified the endogenous function of NOD1. Lymphopoiesis was found to be normal when WT BM cells reconstituted NOD1-/- hosts, whereas transplantation of NOD1-/- BM cells exhibited reduced reconstitution potential compared to WT BM cells, suggesting that NOD1-/- BM cells had intrinsic defects. This finding was unexpected, as the microbial NOD1 ligand promotes hematopoiesis indirectly via mesenchymal stromal cell cytokine secretion, whereas hematopoietic precursors are unresponsive to direct ligand stimulation in vitro. Importantly, NOD1-/- mice did not exhibit decreased serum hematopoietic cytokine levels, indicating that cytokine insufficiency was not responsible for lymphoid defects under homeostatic conditions. Thus, these observations suggested a microbial ligand-independent role for NOD1 in lymphopoiesis.
Given that multiple hematopoietic cytokines converge on STAT5 -mediated signaling, we hypothesized that NOD1 deficiency impairs cytokine responsiveness. Indeed, NOD1-/- HSCs, CLPs, and peripheral CD4+ T cells displayed reduced proliferation in response to hematopoietic cytokines. Correspondingly, the phosphorylation of STAT5 was attenuated in NOD1-/- cells after cytokine stimulation. To mechanistically link NOD1 and STAT5, proximity ligation assays demonstrated that NOD1 and STAT5 physically associate within the cytoplasma of human primary CD4+ T cells in response to cytokines. Notably, this association occurred independently of NOD1 ligand stimulation, suggesting that STAT5 may be a preferred endogenous binding partner of NOD1. Functionally, NOD1-/- HSPCs and CD4+ T cells exhibited a poor survival and proliferation compared to their WT counterparts, correlating with defective STAT5 signaling.
The physiological relevance of these defects was tested by infection with Toxoplasma gondii, a protozoa lacking NOD1 ligands. NOD1-/- mice succumbed more rapidly and harbored a higher parasite burden. Despite normal IL-12 p40 induction, these animals displayed reduced IFN-γ levels, lower CD4+ T cell numbers in the spleen and peritoneal fluids, and impaired expansion of BM HSCs and CLPs. These findings confirm that the absence of NOD1 compromises adaptive immunity through defective lymphopoiesis and the T cell function, independent of microbial ligand recognition (Fig. 1, Lower panels).
A major conclusion of this study is that NOD1 serves dual roles: as a classical pattern recognition receptor that senses bacterial peptidoglycans, and as a microbial ligand-independent regulator of lymphopoiesis via STAT5 signaling. From an evolutionary perspective, NOD1 initially functioned as an innate immune sensor in plants and invertebrates. With th e emergence of the adaptive immune system, it is tempting to speculate that NOD1 plays an additional role as a critical molecule that contributes to the development and maintenance of adaptive immunity.
The rapid spread of SARS-CoV-2 in 2020 highlights the urgent need to understand why some patients develop severe or fatal disease. To address this, our research group initiated a large collaborative study involving seven hospitals in Chiba Prefecture and four university hospitals to clarify mechanisms underlying severe COVID-19[6].
Our initial studies of autopsy samples from fatal cases revealed profound vascular pathology in the lungs. Immunohistochemistry demonstrated SARS-CoV-2 spike protein within the arterial walls, while scanning electron microscope analysis confirmed viral particles localized to the tunica media. Elastin van Gieson staining showed disruption and thickening of elastic fibers accompanied by exudative inflammation in the tunica adventitia and bronchovascular spaces. These findings established that SARS-CoV-2 infection directly induces exudative vasculitis in the pulmonary arteries.
To explore systemic immune dysregulation, single-cell RNA sequencing was performed on peripheral blood mononuclear cells (PBMCs) from COVID-19 patients. Fatal cases displayed a striking reduction in lymphoid populations such as CD4+ T cells and B cells, with concomitant expansion of myeloid clusters, particularly canonical and noncanonical monocytes. A pathway analysis of PBMCs revealed enrichment of the “neutrophil degranulation” and “platelet activation” pathways in fatal cases. Within myeloid subsets, thrombospondin-1 (THBS1) , a molecule that promotes platelet aggregation, has emerged as one of the most highly expressed genes in noncanonical monocytes. A histological analysis confirmed the infiltration of THBS1+ noncanonical monocytes around inflamed pulmonary vessels in fatal cases. Indeed, microthrombi were detected within the infected arteries of the fatal COVID-19 lungs. These observations indicate a mechanism by which noncanonical monocytes contribute to microthrombosis at sites of vascular injury.
Further investigations focused on myosin light chain 9 (MYL9) , a regulatory molecule released from activated platelets[9]. MYL9 has previously been implicated in thrombus-like structures in allergic and inflammatory diseases[9-12]. In patients with fatal COVID-19, MYL9/12 deposition was observed within microthrombi together with platelets, supporting the idea that platelet activation caused by vascular injury induces MYL9 release[6]. Plasma MYL9 levels were assessed in COVID-19 patients. At admission, patients had significantly higher plasma MYL9 concentrations than healthy controls, whereas patients with sepsis or major cardiac surgery did not show such increases. Importantly, elevated plasma MYL9 was found to be correlated with disease severity; severe and critical patients exhibited higher levels than moderate cases, independent of age or sex. Plasma MYL9 levels were correlated with hospitalization duration and predicted oxygen requirement within seven days after admission.
These findings establish Myl9 as both a biomarker and potential effector molecule in COVID-19 pathogenesis.
In summary, severe COVID-19 is characterized by pulmonary exudative vasculitis with direct vascular infection by SARS-CoV-2, infiltration of THBS1-expressing noncanonical monocytes, and the formation of MYL9-containing microthrombi (Fig. 2, Left panels) . Platelet activation induced by vascular injury and THBS1 signaling appears to play a pivotal role in the pathogenesis of severe COVID-19. Measurement of plasma MYL9 levels at hospital admission provides a predictive biomarker for disease severity, clinical course, and therapeutic needs. Beyond its diagnostic utility, targeting MYL9-mediated platelet activation may represent a novel therapeutic strategy to prevent microthrombosis and vasculitis in patients with COVID-19. This integrated pathological and immunological framework advances our mechanistic understanding of severe COVID-19 and offers new avenues for clinical intervention.
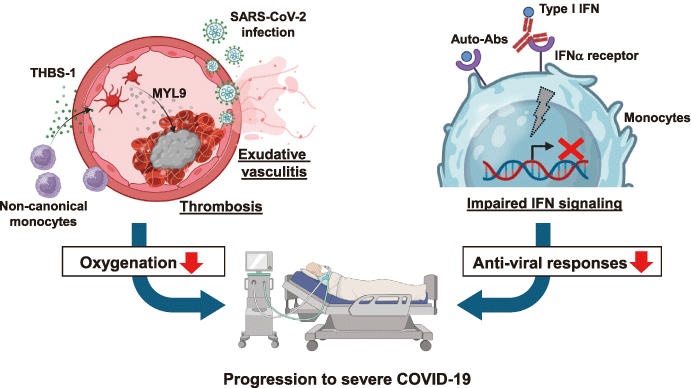
Fig. 2 Mechanisms underlying COVID-19 severity.
SARS-CoV-2 infects the lung blood vessels, leading to exudative vasculitis (left) . This damage recruits non-canonical monocytes that produce thrombospondin-1 (THBS-1) and promote platelet activation. Activated platelets release MYL9, which is deposited in thrombi, and its serum levels are correlated with the disease severity (left) . Reportedly, 10% of severe COVID-19 patients have autoantibodies (auto-abs) against type I interferon (IFN) (right) . These auto-abs can inhibit the binding of IFN-a to its receptor, leading to impaired IFN signaling.
Type I IFNs play a central role in early innate immune defense against SARS-CoV-2. Defects in IFN signaling, whether genetic or acquired, have been linked to life-threatening COVID-19[13-15]. Approximately 10% of such severe cases harbor neutralizing autoantibodies (auto-abs) against type I IFNs, underscoring their contribution to disease severity [16-24]. Our group has confirmed this association and sought to clarify how these auto-abs suppress IFN signaling and alter immune responses [7].
To dissect their impact, we performed single-cell RNA sequencing (scRNA-seq) of PBMCs from COVID-19 patients with anti-type I IFN auto-abs. In patients without auto-abs, critical illness was found to be correlated with robust induction of type I and II IFN-stimulated genes (ISGs) . In contrast, severe or critical patients with auto-abs failed to upregulate ISGs. Myeloid cells, including canonical monocytes and conventional dendritic cells, exhibit markedly reduced expression of antiviral ISGs, indicating that auto-abs blunt type I IFN signaling and impair innate antiviral defense.
Next, we examined B cell responses. An scRNA-seq analysis of BCR repertoires revealed smaller clone sizes and reduced diversity across the IGH, IGK, and IGL chains in patients with auto-abs compared to other groups. Conversion of CDR3 sequences into amino acid profiles confirmed diminished repertoire diversity. Furthermore, a comparison with published datasets showed that SARS-CoV-2-specific clones were underrepresented in auto-abs-positive patients. Consistently, the plasma levels of spike protein-specific antibodies were lower in these individuals than in patients without auto-abs. These findings suggest that impaired IFN signaling compromises the quality of B cell responses and reduces the generation of virus-specific antibodies.
To explore the molecular basis of IFN neutralization, we mapped epitopes of IFN-α2 recognized by patient auto-abs. Plasma samples showed strong reactivity to IFN-α2 peptides spanning residues 80-95 and 124-143, which are regions that overlap with IFNAR1 binding sites. Thus, these auto-abs may directly block IFN-α2 interaction with its receptor, thereby inhibiting downstream signaling.
Clinically, patients with anti-type I IFN auto-abs do not present with distinctive features that would allow identification without laboratory testing. Neutralizing antibody titers must be measured to confirm their presence. Screening for anti-type I IFN auto-abs could identify individuals at higher risk for severe outcomes not only in SARS-CoV-2 infection but also in future viral outbreaks. Proactive identification of carriers and tailored interventions may help mitigate disease severity.
In summary, neutralizing auto-abs to type I IFNs are a key pathogenic factor in severe COVID-19 (Fig. 2, Right panels) . By attenuating antiviral responses in myeloid cells and impairing B cell repertoire diversity and antibody production, these auto-abs compromise both innate and adaptive immunity. Epitope mapping indicates that they act by blocking IFNAR1 binding sites. Measurement of plasma auto-ab titers is essential for the diagnosis, and large-scale screening may help guide preventive and therapeutic strategies against current and future viral infections.
The COVID-19 pandemic later made it clear that in today’s globalized world, infectious diseases that arise overseas can quickly enter and spread within Japan. Although the impact of COVID-19 on society has largely subsided, SARS-CoV-2 continues to mutate, and it remains possible that it could acquire stronger pathogenicity again. In addition, future pandemics caused by other pathogens are realistic possibilities. Therefore, it is essential to recognize them as real threats and to strengthen both research and preparedness for future outbreaks. Simultaneously, the COVID-19 pandemic has emphasized both the success and limitations of mRNA vaccines, including issues of safety, cost, and accessibility. To better prepare for future outbreaks, there is an urgent need for safer, more affordable, and easier vaccines. Since mucosal antigen delivery via tissues such as the gastrointestinal tract can induce strong immune responses, our group, in collaboration with cSIMVa, is a pioneering novel mucosal vaccine platform. These efforts aimed to establish next-generation vaccines from Chiba with global reach.
Not applicable.
I declare that these studies were conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Not applicable.
Not applicable.
I thank Dr. Kiyoshi Hirahara for critically reading this manuscript. I would also like to express my sincere gratitude to Drs. Alan Sher and Dragana Jankovic at the Laboratory Parasitic Diseases of the US National Institute of Allergy and Infectious Diseases and Dr. Toshinori Nakayama (former president of Chiba University) . Thank you to the Chiba Medical Society for awarding me the 17th Chiba Medical Society Award.
Address correspondence to Dr. Chiaki Iwamura.
Department of Immunology, Graduate School of Medicine,
Chiba University, 1-8-1, Inohana, Chuou-ku, Chiba 260-8670,
Japan.
Phone: +81-43-226-2080.
Fax: +81-43-227-1498.
E-mail: iwamurac@chiba-u.jp
