




Chiba Medical J. 92E:15~24,2016
doi:10.20776/S03035476-92E-2-P15
[ The Chiba Medical Society Award(2015) ]
Eizo Watanabe1), Takehiko Oami1), Satoshi Sunahara1), Tomonori Kimura1)
Waka Takahashi1), Masahiko Hatano2) and Shigeto Oda1)
1) Department of Emergency and Critical Care Medicine, Graduate School of Medicine, Chiba University, Chiba 260-8670.
2) Biomedical Research Center, Chiba University Graduate School of Medicine, Chiba 260-8670.
(Accepted February 12, 2016)
Multiple organ failure resulting from severe sepsis is explained as the sum of dysfunction of cells constituting individual organs. Different types of cell death as the final result of cellular dysfunction have been proposed based on their morphological features. In septic cell death, necrosis (cell disintegration associated with massive damage to neighboring cells) is prominent, while apoptosis (type I programmed cell death characterized by an orderly sequence of events) and subsequently autophagy (type II programmed cell death) have been attracting recent research interest. Autophagy (meaning “to eat oneself”) is primarily a mechanism for intracellular proteolytic degradation. Autophagy plays a role in not only nutrition supply as a starvation response but also degradation of non-essential organelles, elimination of pathogenic microorganisms, and tumor suppression, indicating that autophagy is an essential system for maintaining life. However, excessive autophagy induces cell death. The present article reviews the involvement of apoptosis and autophagy (two cell death processes that may potentially work as a two-edged blade depending on the patient’s condition and disease stage) in the pathology of sepsis and discusses the feasibility of treating sepsis by controlling autophagy.
Autophagy, Apoptosis, mitophagy, CLP, IRGM
Multiple organ failure resulting from severe sepsis is explained as the sum of dysfunction of cells constituting individual organs[1]. To date, various types of cell death as the final result of cellular dysfunction have been proposed. Necrosis, passive cell death, is prominent in cases of excessive pathogen invasion and has long been understood morphologically. However, since the dramatic discovery of the role of apoptosis, type I programmed cell death, in the pathology of sepsis[2], the focus has substantially shifted from necrosis to apoptosis in investigations of the pathophysiology of sepsis. The present article reviews the involvement of apoptosis in the pathology of severe sepsis and describes countermeasures against apoptosis in this condition. The findings regarding the involvement of autophagy, type II programmed cell death, obtained to date are also reviewed, and the feasibility of treating sepsis by controlling autophagy is discussed.
Cell death is morphologically classified into three types, necrosis, apoptosis, and autophagy. While the process of apoptosis (programmed cell death) mediated by a protease caspase has been clarified by molecular biological methodologies, necrosis has been considered an accidental (non-programmed) form of cell death. In necrosis, stimuli inducing cell death trigger the influx of calcium ions and activation of calpain. As plasma membrane channels are opened, the influx of extracellular fluid and leakage of lysosomal contents promote intracellular degradation process[3]. As a consequence, both the cytoplasm and nucleus disintegrate, and the cellular injury affects the entire tissue. The extracellular release of the nuclear DNA-binding protein high mobility group box 1 (HMGB1) from the disintegrated nucleus induces further inflammation.
In contrast, apoptosis is the form of physiological cell death that occurs in most cases, and it is the type of cell death associated with embryogenic morphogenesis and postnatal cell renewal of individual tissues. The apoptotic process starts with cell shrinkage, followed by nuclear compaction and loss of mitochondrial membrane potential (Figure 1). Then, further cell shrinkage occurs, with plasma membrane blebbing and formation of apoptotic bodies. In parallel, there is nuclear fragmentation together with chromatin condensation. Finally, the shrunken cells and apoptotic bodies are phagocytized by neighboring cells such as macrophages.
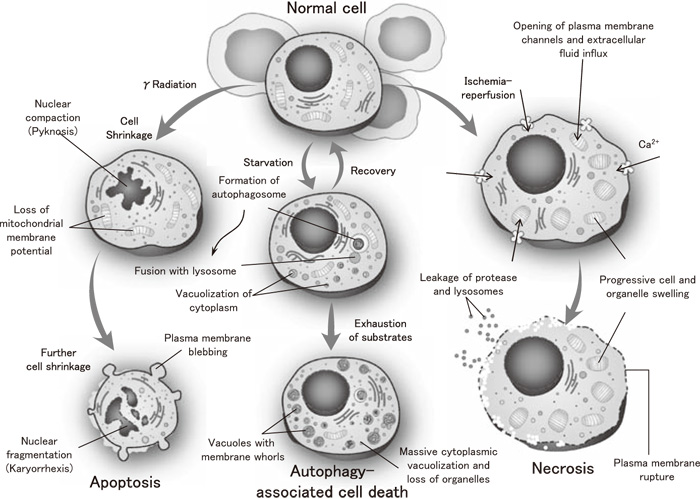
Fig.1
Three pathways of cell death Modified from ref.[3]with permission.
The third type of cell death, autophagy, has recently gained increased attention from researchers. Autophagy is defined as the mechanism by which cell components are transferred to lysosomes (organelles responsible for intracellular digestion) within the same cell and degraded, and the degradation products, such as amino acids and fatty acids, are reused. This internal degradation within a cell constitutes the reason why this process is named “autophagy,” a term derived from the Greek “to eat” (“phagy”) “oneself” (“auto”). The roles of autophagy include not only nutrition supply as a starvation response[4]but also recently identified roles such as degradation of non-essential organelles, elimination of pathogenic microorganisms, and tumor suppression, indicating that autophagy is a system that is essential to supporting life[5].
On the other hand, excessive autophagy results in the enormous loss of organelles due to massive cytoplasmic vacuolization and induces cell death (Figure 1). Autophagy has long been morphologically understood. According to Clarke[6], cell death is classified as follows:
More recently, a new type of cell death, programmed necrosis (necroptosis), induced by the activation of tumor necrosis factor receptor 1 (TNFR1) was proposed[7]. The involvement of RIP kinase 3-dependent necrosis in the lethality of cecal ligation and puncture-induced sepsis in mice has been reported[8].
The landmark study on the involvement of cell death in sepsis may be that by Hotchkiss and colleagues, who were the first to use organ specimens obtained at autopsy from patients with severe sepsis[9]. They found that, among the cells in the various vital organs of the septic patients, apoptotic cell death was observed more frequently than necrosis in lymphocytes and intestinal epithelial cells[9]. They further proposed that marked apoptosis might occur in splenic CD4+ T cells in patients with severe sepsis and lead to acquired immune deficiency syndrome (AIDS)-like immunosuppression[10]. Since this immunosuppression could induce immune paralysis and negatively impact the outcome of severe sepsis, this group has been working to alleviate the paralysis by adopting an approach to control apoptosis in immunocompetent cells[11-13]. A phase 1b/2a trial of anti-programmed death-ligand 1 (PD-L1) antibody, an immune checkpoint inhibitor, in severe sepsis/septic shock is currently in progress in the United States (#BMS-936559: https://clinicaltrials.gov/, accessed 31 January 2016), and its extension to Japan and Europe is planned depending on the results. The clinical usefulness of programmed death 1 (PD-1) expression on leukocytes for monitoring immune dysfunction in critically ill patients has been demonstrated[14], suggesting that this parameter as well as absolute lymphocyte count (ALC) may serve as useful biomarkers for determining the therapeutic effect of treatments for this condition. Furthermore, as the intestinal tract harbors many immunocompetent cells, enhanced apoptosis in intestinal epithelium and reduced proliferation of intestinal crypt cells have been reported in sepsis[15]. Administration of epidermal growth factor (EGF) in sepsis is thought to reduce apoptosis and contribute to suppressing cell death, cell proliferation, and cell migration[16].
On the other hand, persistent organ dysfunction induced by continued mediator production by activated neutrophils due to their delayed apoptosis in sepsis is problematic[17,18]. Accordingly, the different behaviors of different cells should be taken into consideration when selecting countermeasures against apoptosis in sepsis.
We have reported that genetic polymorphisms influence the pathology of conditions such as sepsis during the acute stage[20-23]. To investigate programmed cell death-related genetic polymorphisms, a genotyping study using a commercial single-nucleotide polymorphism (SNP) chip capable of analyzing 2,100 genes and 48,742 SNPs at one time was carried using samples from acute kidney injury, a frequent complication of sepsis[24]. The results demonstrated that minor alleles of two SNPs in the BCL2 gene, an anti-apoptotic gene, did not influence the outcome of sepsis but reduced the risk of developing acute kidney injury secondary to infection (rs8094315: odds ratio 0.61, P=0.0002; rs12457893: odds ratio 0.67, P=0.0002). Another SNP in the SERPINA4 gene linked to the apoptotic pathway in the kidney was found to influence the development of septic kidney injury[24]. These findings were confirmed in multiple Caucasian cohorts. The P2X7 receptor belongs to a family of ligand-gated ion channels activated by extracellular adenosine 5-triphosphate (ATP), which is released in excessive amounts under certain pathological conditions. This receptor is regarded as a “death receptor” that mediates the induction of apoptosis in various cells, and it is also involved in the activation of inflammatory responses. A number of coding SNPs have been identified in the P2X7 gene, and an ex vivo investigation of their influence on ATP-stimulated cytokine production is in progress[25].
On the other hand, the human immunity-related GTPase family M protein (IRGM) gene has been found to play an important role in the autophagic degradation of Mycobacterium bovis (BCG) in cultured human macrophages[26,27]. In addition, a number of IRGM SNPs (e.g., rs13361189, rs10065172) are thought to be involved in excessive inflammatory responses such as those observed in Crohn’s disease[28]. Therefore, genetic polymorphisms of IRGM SNPs were analyzed in 793 ICU patients using an SNP chip in a multicenter study conducted at ICUs in five tertiary emergency medical centers in Japan. The results demonstrated the involvement of the TT genotype of the SNP locus IRGM (+313) (rs10065172) in the poor outcomes of patients with severe sepsis (Figure 2) and relatively low expression levels of IRGM mRNA caused by lipopolysaccharide (LPS). This suggests the possible role of a decreased autophagic response to a septic insult in poorer outcomes.
Before reviewing involvement of autophagy in sepsis, the types, membrane dynamics, and molecular mechanism of autophagy will be outlined.
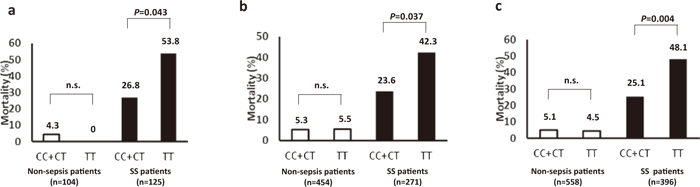
Fig.2
Comparison of the mortality between different genotype categories of the SNP at IRGM( +313)(rs10065172)[54].(a) The discovery cohort(P=0.043, recessive model of the correlation/trend test; TT v(CC+CT) in 125 SS patients).(b) The multi-center validation cohort(P=0.037, recessive model of the correlation/trend test; TT v(CC+CT) in 271 SS patients).(c) The combined cohort( P=0.004, recessive model of the correlation/trend test; TT v(CC+CT) in 396 SS patients). SS, severe sepsis/septic shock
At least three different types of autophagy exist in mammalian cells[19]: (1) macroautophagy, (2) chaperon-mediated autophagy (CMA), and (3) microautophagy.
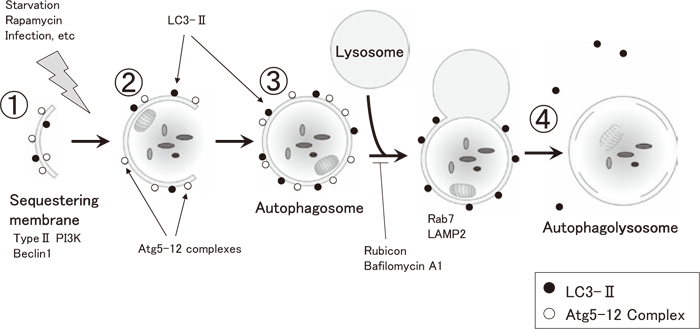
In macroautophagy, substances and organelles are first surrounded by a membrane called a “sequestering membrane.” This membrane appears in the cytosol and fuses into a double-membrane vesicle known as an autophagosome, which surrounds the substrates to be degraded. Then, an autophagosome fuses with a lysosome.
In CMA, substrates to be degraded are recognized by a molecular chaperon, heat shock protein 70 (hsp70), in the cytosol and directly pass through the lysosomal membrane for degradation.
Microautophagy involves the direct trapping of organelles by the lysosomal membrane for subsequent degradation.
Among these three types of autophagy, macroautophagy exhibits the highest activity of proteolytic degradation and is capable of sequestering and degrading a wide variety of substrates, from protein molecules to organelles, in autophagosomes with a diameter of approximately 1 μm. Therefore, this process is a bulk degradation system, and the term “autophagy” generally refers to macroautophagy. Although another intracellular protein degradation system, the ubiquitin-proteasome system, consists of a large protein complex, autophagy involves membrane trafficking mediated by membrane dynamics.
When cells are exposed to stresses such as starvation, the autophagic mechanism is activated by type III phosphatidylinositol (PI) 3-kinase/Beclin 1 (Atg6) and other factors (Figure 3). Multiple steps are involved in the membrane dynamics of autophagy: ①spontaneous formation of the sequestering membrane; ②extension of the sequestering membrane in the presence of Atg5-12 and Atg16 complexes; ③fusion of the sequestering membrane to form a double-membrane autophagosome with a vesicle-like structure; and ④fusion of the outer membrane of the autophagosome with a lysosome to form an autophagolysosome and subsequently degrade the contents. Atg5-12 complexes (represented by the symbol ○ in Figure 3) dissociate from the membrane surface following autophagosome formation. On the other hand, LC3-II (Atg8-phosphatidyl ethanolamine (PE) complex) (represented by the symbol ● in Figure 3) binds to the sequestering membrane in an Atg5-12 complex-dependent manner and contributes to autophagosome formation. In contrast to the ubiquitin-proteasome system described above, this system degrades bulk substrates such as proteins without any specific recognition process and is capable of degrading/digesting organelles such as mitochondria (mitophagy) (see below).
We hypothesized that autophagy, type II programmed cell death, might play an important role in the pathology of sepsis and performed an electron microscopic morphological investigation of autophagosomes in liver samples obtained from patients with severe sepsis[29]. The results demonstrated that there were significantly more autophagosomes in patients with severe sepsis than in control patients without sepsis[29]. This phenomenon was reproduced in liver tissue specimens obtained from mice with surgical sepsis induced by cecal ligation and puncture (CLP) 24 hours after surgery, which showed an increased number of autophagosomes in the liver during sepsis[29]. This is the first article documenting the involvement of autophagy in severe sepsis. To clarify whether the observed increase in autophagosomes indicated an enhancement of autophagy or resulted from the interruption of the autophagic process, further studies of autophagy flux were performed using liver samples obtained from CLP-mice. The results demonstrated a marked increase in the number of autophagic structures at an even earlier stage, 6 hours after operation (Figure 4). Furthermore, an increase in the amount of p62 protein over time, reflecting the accumulation of autophagosomes, indicated the transient enhancement of autophagy flux in response to the septic insult, although there was a tendency for subsequent stagnation[30]. We recently observed a similar tendency in murine proximal tubular cells as well (unpublished data), consistent with a previously reported increase in autophagosomes in specimens obtained at autopsy from septic patients[31]. In the murine sepsis model described above, inhibition of autophagy with chloroquine resulted in deterioration of liver function and an increase in the mortality rate[30], while stimulation of autophagy with rapamycin normalized the serum cystatin C level, a marker of renal dysfunction (unpublished data). These findings suggested the transient enhancement of autophagy in these organs as a part of the biological defense during the subacute stage of sepsis.
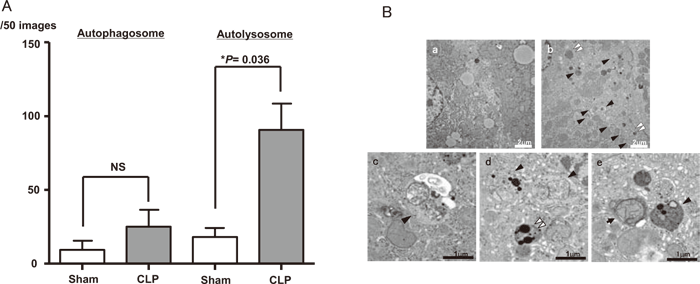
Fig.4
(A) The number of autophagosomes and autolysosomes are compared between CLP and sham animals.All data are expressed as the mean±SD. Data were analyzed for statistical significance using the MannWhitney test. The increase in autolysosomes in the CLP group was statistically significant(*P<0.05; n=3); the mean increases in autophagosomes in the CLP group compared to the sham group did not reach statistical significance. NS, not significant.(B) Electron microscopic images of the liver; a: Liver sample obtained from sham-operated mice. Organelles in the hepatocyte are generally intact, and lysosomes do not contain discrete membrane structures, although the non-homogeneous electron-dense material often seen in(hetero) lysosomes most certainly represents the end-stage degradation of phospholipids and other cytoplasmic materials(material at the light microscopic level referred to as lipofuscin); b-e: CLP-operated mice. Double arrow heads indicate the complex structures bounded by two membranes(autophagosomes); arrow heads indicate single membrane-bound lysosomal complexes with degraded organelle content(autolysosomes); e: the double arrow head indicates an autophagosome that clearly contains a damaged mitochondrion.
While autophagy has attracted the attention of researchers due to its role in innate immune responses against pathogenic microorganisms[32-35], a portion of endogenous Epstein-Barr virus nuclear antigen 1 (EBNA1) following degradation in autophagolysosomes plays an important role in antigen presentation to CD4+ T cells via major histocompatibility complex (MHC) class II molecules[36]. Similarly, for the process of antigen presentation to CD8+ T cells via MHC class I molecules, the accumulation of viral antigen proteins in autophagosomes during herpes simplex virus (HSV)-1 infection indicates the importance of autophagy in antigen processing[37]. In addition, deficiency of autophagy-related gene 5 (Atg5) was found to cause abnormalities in self-antigen presentation by MHC class II molecules on thymic epithelial cells[38]. Furthermore, an autophagic mechanism has been reported to be critical in the differentiation and survival of T lymphocytes[39]. Based on these findings, autophagy likely has many more roles in immune mechanisms than previously thought.
As described earlier, the involvement of the apoptosis (type I programmed cell death) of immunocompetent cells in sepsis has almost been proven[2,9,10]. Accordingly, the degree of autophagy (type II programmed cell death) was investigated in CD4+ T cells exhibiting markedly enhanced apoptosis in sepsis.
First, when sepsis was induced by a CLP operation in GFP-LC3 mice containing fluorescence-labeled autophagosomes throughout their whole bodies[30,40], a significant increase in the expression of LC3 in CD4+ T cells was observed 24 hours after surgery, demonstrating an increase in autophagic structures. However, an increase in lysosomes and the accumulation of p62 were also observed in this animal model, which suggested the possibility that autophagy in sepsis might not be sufficient for biological defense. On the other hand, following a CLP operation in Atg5 conditional knockout mice (CD4-Cre/Atg5f/f) specifically lacking autophagy in T cells, a significant decrease in the number of CD4+ T cells was observed at an early stage after the CLP surgery with a concomitant enhancement of apoptotic activity compared with the sham-operated animals (Oami, et al. submitted to “Critical Care Medicine”).
During the process of tissue dysoxia in severe sepsis, reactive oxygen species (ROS) are generated via activation of nuclear factor-kappa B (NF-κB), and cells are exposed to substantial oxidative stress. ROS are also produced as byproducts of mitochondrial electron transport for ATP synthesis by the catalytic activities of enzymes, including NADPH-dependent oxidase (Nox). The generated ROS damage organelles and DNA. On the other hand, since mitochondria have anti-oxidative mechanisms to eliminate ROS, oxidative stress is closely related to the quality control of damaged mitochondria that produce ROS. Furthermore, damaged mitochondria release cytochrome C into the cytoplasm, which induces apoptosis via the intrinsic pathway. Mitophagy is a process used for the selective autophagic removal of dysfunctional mitochondria[41], and in the field of response to insult, there has been recent clinical and research interest in the effects of mitophagy in liver cells and skeletal muscle cells on the pathology of sepsis[29,42-44]. In clinical cases, activation of mitochondrial biogenesis in the skeletal muscle in septic patients is reported to contribute to a favorable outcome[43], which implies that quality control of mitochondria by autophagy may play an important role in this condition[45]. We observed the accumulation of mitochondria in CD4+ T cells in the CD4-Cre/Atg5f/f mice described above (Oami, et al. submitted to “Critical Care Medicine”), indicating the accumulation of dysfunctional mitochondria in a septic insult.
According to a number of reports, inhibition of autophagy triggers apoptosis in cells[46,47]. Atg5 is cleaved by calpain, a protease that triggers necrosis, and induces apoptosis by binding to Bcl-xL, an anti-apoptotic transmembrane protein component of mitochondria[48]. It is also known that the anti-apoptotic Bcl2/Bcl-xL complex conjugates with Beclin 1 to inhibit autophagy[49]. Thus, studies to clarify the cross-talk between the different mechanisms of cell death at various levels are currently in progress. We observed enhanced expression of PDCD1, a pro-apoptotic gene, a significant reduction in BCL2, an anti-apoptotic gene, and enhanced apoptotic activity in CD4+ T cells in CD4-Cre/Atg5f/f mice (Oami, et al. submitted to “Critical Care Medicine”). While mRNA expression in a model of an acute disease such as sepsis greatly varies over time and depends on cell type[50], we propose that substantial cross-talk may exist between autophagy and apoptosis upon the initiation of these two processes.
As described above, the close involvement of autophagy during the onset of organ dysfunction in sepsis has been clarified. Accordingly, controlling autophagy is expected to be an effective measure to prevent or treat organ dysfunction in sepsis. There are several possible methods for controlling autophagy. For example, autophagy is known to be induced not only by starvation stress but also by drugs such as rapamycin. On the other hand, biomolecules such as insulin and growth factors inhibit autophagy by activation of mammalian target of rapamycin (mTOR) via a downstream pathway. The drug bafilomycin A1 is also reported to inhibit autophagy by inhibiting the fusion of autophagosomes with lysosomes[51]. In addition, the harmful effects of overfeeding in acute conditions have long been concerned[52], and the concept of permissive underfeeding has been widely recognized[53]. These facts imply that maintaining moderate nutrient starvation state to avoid autophagy suppression may potentially contribute to improvement of clinical conditions in severe sepsis.
Sepsis is a complex syndrome that can be triggered by infection by any pathogen and encompasses a wide variety of pathological conditions. Furthermore, interindividual differences in the immune/inflammatory response are also an important factor[20-23]. To address and overcome sepsis, a tough enemy, it is imperative for us intensivist to carefully consider the patient's immune status, nutritional status, interindividual differences, and disease stage after proper stratification and to not miss the therapeutic window, which can be very narrow. Monitoring and controlling programmed cell death as described in the present article may provide a breakthrough approach that will improve survival in septic patients.
The present work was partly supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Culture of Japan (#23792070, #15K20333, and #15K20335). The first author wishes to thank the mentorship of Profs. Richard S. Hotchkiss (Washington University), Timothy G. Buchman (Emory Center for Critical Care), and Hiroyuki Hirasawa (Chiba University).
Address correspondence to Dr. Eizo Watanabe.
Department of Emergency and Critical Care Medicine, Graduate School of Medicine, Chiba University, 1-8-1, Inohana, Chuou-ku, Chiba, 260-8670 Japan.
Phone: +81-43-226-2372. Fax: 81-43-226-2371.
E-mail: watanabee@faculty.chiba-u.jp
