




Chiba Medical J. 93E:63~68,2017
doi:10.20776/S03035476-93E-5-P63
[ The Chiba Medical Society Award(2016) ]
Kiyoshi Hirahara
Department of Immunology, Graduate School of Medicine, Chiba University, Chiba 260-8670.
(Accepted August 17, 2017)
CD4+ T cells are crucial for directing appropriate immune responses during the host defense and for the pathogenesis of inflammatory diseases through their functional diversification. Transcription factors and cytokines play crucial roles in the generation and maintenance of the functional diversity of CD4+ T cells. In the present review, I focus on how these various factors contribute to sharpen the function of CD4+ T cells. In particular, I focus on the transcription factor BACH2, which is associated with various autoimmune diseases. I will also discuss our recent findings with regard to the immunosuppressive cytokine, IL-27 and its activated signal transducer and activator of transcription(STAT) 1 and STAT3. Understanding the precise mechanisms of CD4+ T cell-induced immune hemostasis will become increasingly important for overcoming intractable immune-mediated inflammatory diseases.
immune homeostasis, Bach2, STAT1, STAT3, IL-27, IL-6, Immune homeostasis, CD4+ T cells
CD4+ T cells are a key cell population that shapes appropriate adaptive immune responses during the host defense against micro-invaders. While, CD4+ T cells may also play pathogenic roles as drivers of various immune-related diseases including asthma. The diversity of CD4+ T cell subsets including Th1, Th2, Th17, Th9, Th22, T follicular helper cells and regulatory T cells is now well appreciated[1,2]. Transcription factors play key roles in the generation of these diverse subsets and the exertion of the appropriate function by each subset. In particular, negative regulators often act in conjunction with positive factors to stabilize specification. Bach2 was originally reported to be a negative regulator of the differentiation of plasma cells[3]. Polymorphisms within a locus encoding the transcription factor BACH2 have been reported to be associated with diverse immune-mediated diseases, including asthma, multiple sclerosis, Crohn's disease, coeliac disease, vitiligo and type 1 diabetes[4-8]. More recently, BACH2 haplosufficiency was reported to cause a syndrome of BACH2-related immunodeficiency and autoimmunity (BRIDA)[9]. However, the role of Bach2 in CD4+ T cells has not been established. Recently, we and others reported the crucial roles of Bach2 in T cells[10,11]. With regard to CD8+ T cells, Bach2 has been reported to be crucial in the generation of memory T cells[12]; while in CD4+ T cells, Bach2 stabilizes regulatory T cell formation by repressing the differentiation programs of multiple effector lineages, such as Th1, Th2, and Th17 cells[10]. Bach2 is required for the efficient formation of regulatory T cells and consequently for the suppression of lethal inflammation, as Bach2- deficient mice suffer from spontaneous chronic lung inflammation. Consequently Bach2 maintains immune homeostasis through a non-redundant role in regulatory T cell development. The assessment of the genomewide function of Bach2 revealed that it repressed genes associated with effector T cell differentiation. The genes in regulatory T cells that are directly targeted by Bach2 include Gata3 , Irf4 and Il12 rb1 . As a result, its absence during regulatory T cell formation causes inappropriate diversion to effector lineages and the loss of the regulatory function. In addition, Bach2 constrained full effector differentiation within Th1, Th2 and Th17 cell lineages. Taken together, these findings highlight Bach2 as a key regulator of CD4+ T-cell differentiation that prevents inflammatory disease by controlling the balance between tolerance and immunity(Figure 1) [10]
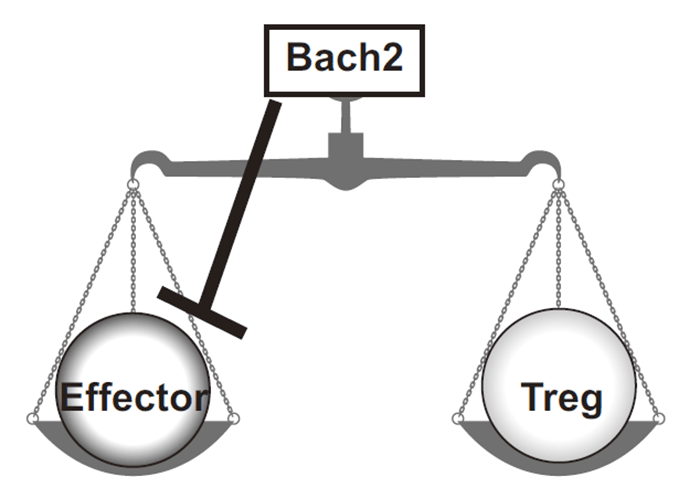
Fig. 1
Bach2 regulates immune homeostasis through controlling the balance between effector T cells and regulatory T cells.
Cytokines are another critical factor in the orchestration of the immune responses to diverse microbial pathogens; they are also major drivers of immune-mediated disease. Many of the most successful new drugs are themselves cytokines or target cytokines [13]. Among the various cytokines, Interleukin (IL)-27 is considered to be a member of a family of heterodimeric cytokines with critical immunoregulatory properties [14,15]. Other heterodimeric cytokines, IL-12 and IL-23, are critical for the differentiation of T helper 1(Th1) and Th17 cells. IL-27 was initially considered to be important for inducing the expression of T-bet and enhancing Th1 differentiation[16]. Later, it became apparent that IL-27 has essential immunosuppressive actions because IL-27 receptor(IL-27R)-deficient mice showed lethal inflammation upon infection with various pathogens(i.e., Trypanosoma cruzi or Toxoplasma gondii). Il27 ra-deficient mice showed severe pathological changes due to enhanced Th1 and Th2 responses during infection with Trypanosoma cruzi[17]. Similarly, infection with Toxoplasma gondii caused uncontrolled tissue damage mediated by activated Th1 cells and increased IL-17 production in Il27 ra-deficient mice[15,18].
The immunosuppressive activity of IL-17 is mediated by various underlying mechanisms. Inflammatory cytokine production in activated T cells can be directly inhibited by IL-27 in vitro[19,20]. In particular, Th17 cell differentiation is diminished by IL-27 through the inhibition of the expression of RORγt, a master transcription factor for Th17 cell differentiation [21]. Consistent with these findings, in a mouse model of brain inflammation-in which Th17 cells play a key pathogenic role-mice deficient in IL- 27R developed more severe Th17 cell-associated neuropathology, whereas the treatment of wild-type mice with recombinant IL-27 could constrain Th17 cell differentiation and abolish the development of experimental autoimmune encephalomyelitis (EAE)[22,23]. In addition to inhibiting Th17 cell differentiation, IL-27 could also enhance the production of an important immunosuppressive cytokine, IL-10, in many helper T cell subsets[21,24,25]. Moreover, IL- 27 has been shown to work together with TGF-β to drive the differentiation of IL-10-producing regulatory type 1 T cells(Tr1) through the induction of aryl hydrocarbon receptor(AhR), c-Maf, IL-21 and ICOS [26-29]. Further support for the immunoregulatory role of IL-27 has been suggested from data linking IL27 polymorphisms with asthma, chronic obstructive pulmonary disease, inflammatory bowel disease and diabetes [30-33].
IL-27 also has diverse effects on non-T cell immune cells. For instance, NK cells produce enhanced IFN-γ by IL-27[34], whereas, IL-27 can suppress the LPSinduced production of cytokines by dendritic cells (DCs) in vitro[35].
Despite its important actions, little had been known in relation to the genes that are directly regulated by IL- 27. We performed RNA-sequencing with naive CD4+ T cells stimulated with IL-27 and analyzed their global gene expression. Cd274 , which encodes PD-L1, was one of the genes upregulated by IL-27. PD-L1 is one of the ligands for programmed cell death protein 1(PD-1) and the PD-1-PD-L1 pathway is recognized to be critical in maintaining peripheral immune tolerance[36-38]. PD-L1-deficient mice show exacerbated disease in EAE, autoimmune arthritis, and autoimmune diabetes, while mice lacking PD-L1 do not develop spontaneous autoimmune disease[36,39-41].
We found that the IL-27-dependent induction of PD-L1 on naïve CD4+ cells inhibited IL-17 production in an untreated population of differentiating Th17 cells and that the development of Th17 cells-mediated autoimmune encephalomyelitis was ameliorated by the adaptive transfer of IL-27-treated naïve CD4+ T cells and that this was accompanied by the high expression of PD-L1. Taken together, we identified a novel mechanism by which IL-27 can exert its ability to suppress IL-17-mediated pathology(Figure 2)[42].
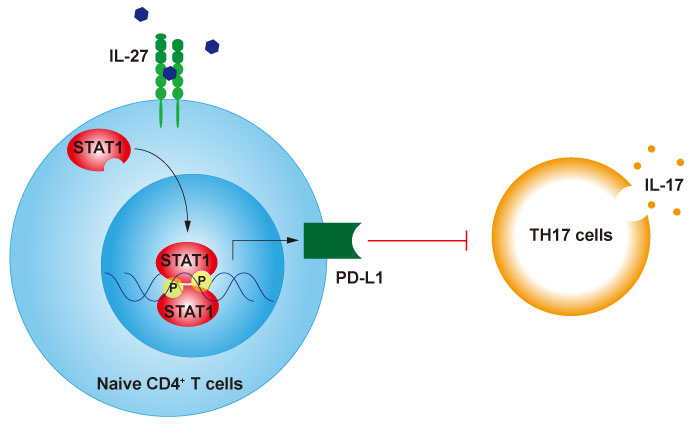
Fig. 2
IL-27 negatively regulates Th17-mediated inflammatory responses through the induction of PD-L1.
As explained above, IL-27 is a novel immunesuppressive cytokine. IL-27 binds to a receptor composed of IL-27R and glycoprotein 130(gp130) [43]. Gp130 is a shared receptor subunit that is also used by other cytokines, including IL-6, which is a type of inflammatory cytokine[44,45]. The sharing of the gp130 subunit between IL-27 and IL-6 is notable because these two cytokines have opposing immunological functions: IL-6 inhibits Th1 cell differentiation, enhances Th2 cell differentiation and induces Th17 cell differentiation. In contrast, IL-27 induces Th1 cell differentiation and inhibits both Th2 and Th17 cell differentiation[46-48]. Like the other type 1 and 2 cytokine receptors, IL-27 and IL-6 activate Janus kinases and signal transducer and activator of transcription(STATs), mainly via a combination of STAT3 and STAT1, and regulate the gene expression[49-52]. The pro-inflammatory effects of IL-6 have been attributed to STAT3, whereas it has been argued that the ability to activate STAT1 explains the" Th1" features of IL-27, as well as its ability to inhibit Th17 cells [53]. However, the" immunosuppressive" functions of IL-27 have also been attributed to STAT3[54]. These previous works imply that the ability of both cytokines to activate both STATs seem paradoxical, given their functions. How do these same STATs control the distinct biological effects of IL-6 or IL-27? A major goal of our study was to understand the molecular underpinnings that defined the specificity and redundancy of the cytokine response through the action of the STAT proteins.
Using a genome-wide approach that permits a comprehensive, quantitative analysis (i.e., RNASeq and ChIP-Seq), we found that IL-6 and IL-27 regulated many of the same genes and thus exhibited substantial redundancy. IL-6 and IL-27 also had discrete transcriptomic profiles despite the commonality in signal transduction. Importantly, we found that the major impact of STAT3 went beyond just IL-6-regulated genes by investigating the gene expression profiles in STAT3- deficient CD4+ T cells. In fact, most genes selectively regulated by IL-27 were also under the control of STAT3, and the overall gene expression level driven by both IL-6 and IL-27 was profoundly STAT3-dependent. In sharp contrast, the major role of STAT1 is to drive distinctive programs that are unique to each cytokine, as evidenced by STAT1-deficient CD4+ T cells. Moreover, the similarity in the IL-6- and IL-27-regulated gene expression was decreased in T cells from patients with STAT1 gain-of-function mutations, which were associated with a rare primary immunodeficiency with fungal infection[55], in comparison to healthy controls. Thus, the results provide a quantitative evaluation of the asymmetric contributions of STAT3 and STAT1, which shape specificity in gp130-mediated cytokine responses(Figure 3)[56]. Our results suggest that the diverse biological effects of cytokines are driven via various combinations of activated STATs and that they contribute to the maintenance of immune homeostasis.
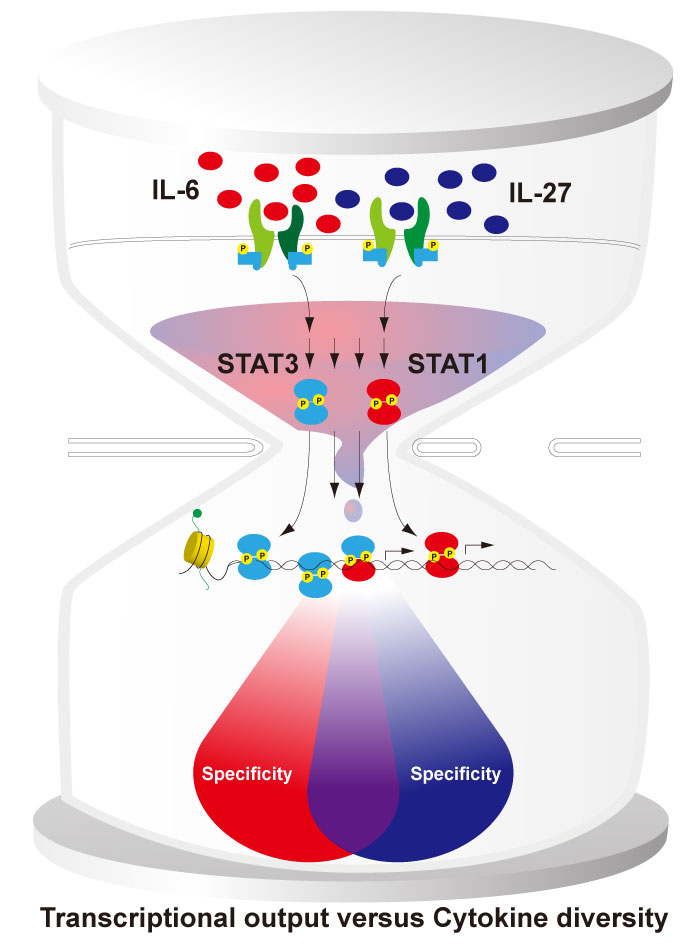
Fig. 3
Asymmetric STAT action in driving IL-27 and IL-6 transcriptional outputs and cytokine specificity.
I have briefly reviewed the recent progress of our research regarding key factors that are involved in the function of CD4+ T cells in relation to the control of immune homeostasis. There is increasing evidence to support the mechanisms through which CD4+ T cells control immune homeostasis. The dysfunction of these mechanisms is involved in various immune-mediated inflammatory diseases. In order to develop new therapeutic strategies for intractable immune-mediated inflammatory diseases, it will therefore become increasingly important for immunologists and clinicians to understand the precise mechanisms of CD4+ T cellinduced immune hemostasis.
I greatly appreciate the continuous help and suggestions from my mentor, Professor Toshinori Nakayama in the Department of Immunology, Graduate School of Medicine at Chiba University. I also appreciate the important help and suggestions from Drs. John O'Shea and Yuka Kanno at the Molecular Immunology and Inflammation Branch of the US National Institute of Arthritis and Musculoskeletal and Skin Diseases. I would like to thank to The Chiba Medical Society for giving me The 8th Chiba Medical Society Award.
Address correspondence to Dr. Kiyoshi Hirahara.
Department of Immunology, Graduate School of Medicine, Chiba University, 1-8-1, Inohana, Chuo-ku, Chiba 260-8670, Japan.
Phone: +81-43-226-2080. Fax: +81-43-227-1498.
E-mail:hiraharak@chiba-u.jp
