




Chiba Medical J. 94E:19-23,2018
doi:10.20776/S03035476-94E-3-P19
[ Chiba Medical Society Award Review ]
Motoko Y. Kimura
Department of Medical Immunology and Department of Immunology Graduate School of Medicine, Chiba University,Chiba 260-8670.
(Accepted March 22, 2018)
The thymus is the only organ that produces T cells where immature thymocytes are properly educated and make the correct decision to become appropriate lineage T cells such as CD4+ or CD8+ T cells. This process is referred to as‘ lineage choice’. Failure in T cell development and/or lineage choice is a direct cause of severe immunodeficiency and/or severe autoimmune disorders. Thus, it is extremely important to understand the molecular mechanisms of thymocyte development and lineage choice and many immunologists have studied this issue intensively.
A diverse TCR repertoire(nearly 1018) is generated by TCR gene rearrangement in immature thymocytes. It contains autoreactive T cells as well as useless T cells that cannot recognize self- MHC molecules. These useless T cells are negatively selected(i.e. clonal deletion), and only cells that moderately recognize self-MHC molecules are positively selected. These processes are termed negative and positive selection. Positively selected cells further differentiate into either CD4T cells or CD8T cells, depending on their specificity to MHC-II or MHC-I respectively. How can the lineage choice of T cells, which have a huge TCR repertoire, be properly determined during their development? In this review, I will update the recent understanding of the molecular mechanisms underlying the CD4/CD8 lineage choices in the thymus and explain the mechanisms of error-free MHC-I-dependent CD8 T cell development.
thymus, lineage choice, T cell development, MHC-I, CD8T cells
The thymus is the only organ that supports T cell development, educates T cells, and produces mature T cells[1]. Early T cell progenitor cells in the thymus, which come from the bone marrow, do not yet have any features of T cells; they neither express CD4/CD8 coreceptors(this is why these cells are named double negative [DN] cells) nor T cell antigen receptors (TCRs) on their surface. DN cells first rearrange their TCRβ chain and form preTCRs together with their preTα chain, and the cells that successfully form preTCRs go through the process called β-selection. β-selection induces massive proliferation, and eventually the cells become CD4+CD8+ doublepositive(DP) cells. DP cells rearrange their TCRα chain and eventually express the complete form of TCRαβ on their cell surface for the first time during their development. TCR rearrangement, the process discovered by Nobel laureate Dr. Susumu Tonegawa, theoretically creates a broad TCR repertoire(nearly 1018). Each DP cell expresses different TCRs with specificity to different antigens. Importantly, DP cells that express the complete form of TCRαβ undergo a further developmental process, depending on their specificity to the self-antigens presented by the MHC class I(MHC-I) or MHC class II(MHC-II) complexes (self-MHCs), which are expressed by the thymus[1] (Fig.1). For instance, DP cells that have a TCR with a strong affinity to self-MHCs undergo clonal deletion by a process called‘ negative selection’; whereas, DP cells that have a TCR with little or no affinity to self- MHCs undergo cell death by a process called‘ death by neglect’. Only DP cells that have a TCR with moderate affinity to self-MHCs are positively selected and proceed to the next developmental step(Fig.1). Positively selected DP cells eventually become either CD4T or CD8 T cells, depending on the specificity of their TCR to the MHC molecule. In this process, called ‘CD4/CD8 lineage choice’, DP cells that express TCRs that recognize the self-MHC-I molecule become CD8T cells, while DP cells that express TCRs that recognize self-MHC-II become CD4 T cells.
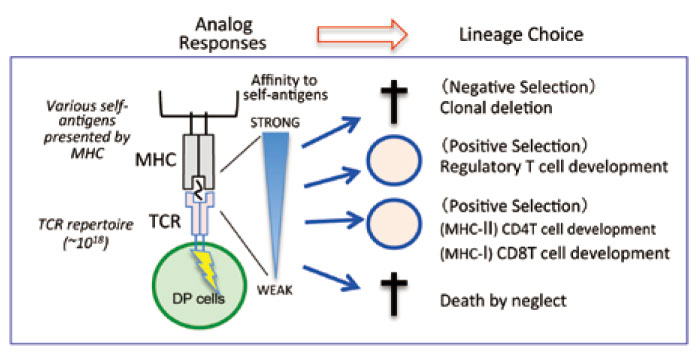
Fig. 1 T cell development in the thymus
Upon TCR gene rearrangement, a broad TCR repertoire (nearly 1018) is created. Each DP cell expresses a unique TCR, and undergoes a selection process to determine its specificity to self-antigens presented by either MHC-I or MHC-II molecules that are mainly expressed on thymic epithelial cells. The reactivity of each TCR to self-MHCs determines the lineage fate.
Considering the fact that DP cells have a huge TCR repertoire and that each DP cell has a chance to interact with‘ various’ self-antigens presented by thymic epithelial cells(TECs), the interaction between TCRs and self-MHCs must create numerous“ analog” responses. However, surprisingly, these analog responses result in a“ unique” lineage choice with almost no errors (Fig.1). This fact suggests that a strict mechanism must exist to allow for an“ error-free” proper lineage choice. A recent study showed how each DP thymocyte expressing different MHC-I-restricted TCRs properly differentiated into CD8 T cells[2]. This mechanism will be explained later in this review.
Many immunologists have extensively studied the mechanisms of the CD4/CD8 lineage choice in the thymus, and many models have been proposed and discussed[3-5]. After decades of intense debate,“ the kinetic signaling model” proposed by the Alfred Singer Laboratory (NCI/NIH) is widely accepted as the mechanism of CD4/CD8 lineage choice[4]. We herein describe the kinetic signaling model(Fig.2), and then introduce the recent studies that further advance the knowledge about the CD4/CD8 lineage choice.
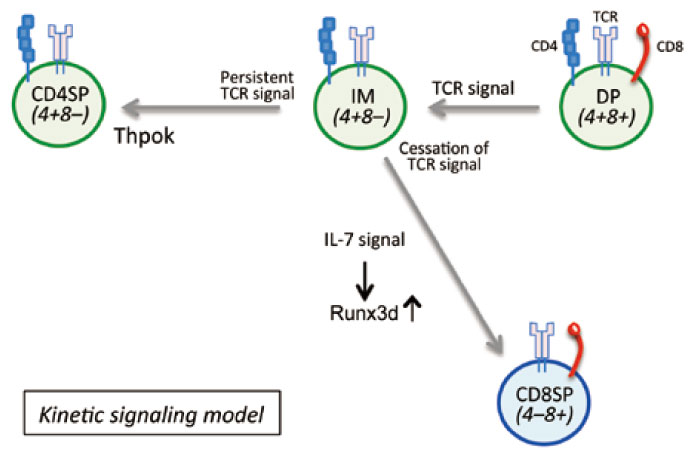
Fig. 2 The kinetic signaling model
TCR-signaled positively selected cells first terminate CD8 gene transcription, and become CD4+CD8low(Cd4+Cd8-) intermediate (IM) cells. Since MHC-I TCR signaling is dependent on the CD8 coreceptor, the loss of the surface CD8 expression results in the cessation of TCR signaling, which allows IM cells to respond to IL-7, inducing the expression of Runx3d, which dictates the CD8 lineage fate. In contrast, the TCR signaling of MHC-IIselected cells is continuous, since MHC-II TCR signaling is independent of the expression of CD8. Persistent TCR signaling induces the expression of Thpok, which results in CD4 T cell development. The kinetic signaling model suggests that the duration of TCR signaling determines the CD4/CD8 lineage choice.
DP thymocytes that express the complete form of TCR start interacting with TECs that express various self-antigens presented by either MHC-I or MHC-II molecules. The successful interaction between TCRs and self-MHCs provides TCR signaling, which initiates the positive selection process. Positively selected cells first terminate CD8 gene transcription, regardless of their TCR specificity to either MHC, and cells become CD4+ CD8low(Cd4+Cd8-) intermediate(IM) cells. Since the MHC-I TCR signal is dependent on the CD8 coreceptor, the loss of the surface CD8 expression results in the cessation of TCR signaling. The cessation of TCR signaling allows IM cells to respond to cytokines such as IL-7[6,7], which induces the expression of Runx3d, dictating the CD8 lineage fate. In contrast, the loss of the surface CD8 expression does not interfere with MHCII- dependent TCR signaling, since the MHC-II TCR signal is independent of the CD8 expression; thus, the TCR signaling of MHC-II-selected cells is continuous. Persistent TCR signaling induces the expression of Thpok, which promotes CD4 T cell development(Fig.2). In summary, the kinetic signaling model suggests that th‘e duration’ of TCR signaling determines the CD4/ CD8 lineage choice. A long duration of TCR signaling induces the expression of Thpok, promoting CD4 helper lineage cells; whereas, a short duration of TCR signaling results in the induction of IL-7-dependent Runx3d, promoting CD8 cytotoxic lineage cells(Fig.2).
Considering the fact that there is a broad TCR repertoire and that there are TECs present various types of self-antigens, the affinity of the interactions between TCRs and self-MHCs in each cell must be very different and these differences are likely to promote different durations of TCR signaling. Nevertheless, all MHC-II-selected cells become CD4T cells and all MHC-I selected cells become CD8 T cells, which may contradict the kinetic signaling model that suggests that the‘ duration’ of TCR signaling determines the CD4/ CD8 lineage choice. How could the lineage choice be well organized[2,4].?
Let us look at the details of the process of MHC-I positive selection(Fig.3). MHC-I-dependent CD8 T cells develop in two steps. In the first step, DP cells are positively selected and become CD4+CD8low IM cells; this step is referred to as“ phase 1”. In the second step, IM cells become CD8T cells; this step is referred to as “phase 2”. Phase 1 cells are“ lineage-undefined” cells; whereas, Phase 2 cells are Runx3d expressing“ CD8 lineage-defined” cells(Fig.3).
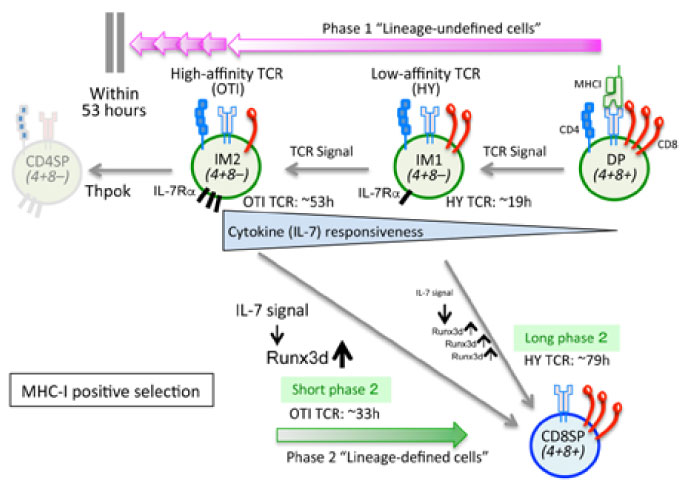
Fig. 3 The basis of MHC-I-dependent positive selection
MHC-I-dependent CD8 T cell development has two steps. In the first step, positively selected DP cells become CD4+CD8low IM cells; this is referred to as“ phase 1”. In the second step, IM cells become CD8T cells by responding cytokines such as IL-7; this is referred to as “phase 2”. Phase 1 cells are“ lineage-undefined” cells; whereas Phase 2 cells are Runx3-expressing“ CD8 lineagedefined” cells. Phase 1 is controlled by TCR signaling; thus, DP cells that express low-affinity TCRs(e.g. HYTCR) have a short phase 1 but long phase 2, while DP cells that express high-affinity TCRs(e.g. OTI-TCR) have long phase 1 but short phase 2. A longer phase 1 duration increases the cytokine responsiveness of IM cells so that the cytokine-driven duration of Phase 2 becomes shorter. As a result, the total time for positive selection is almost constant. Error-free MHC-I positive selection requires phase 1 to end within 53 hours.
To examine the impact of different TCR affinities against self-MHC-I for CD8 T cell development, we utilized two different TCRs: HY-TCR(female), which is known to have low affinity to the self-MHC-I molecule; and OTI-TCR, which is known to have high affinity to the self-MHC-I molecule. To measure the amount of time in each phase, we utilized RAG2 GFP Tg mice[2,8]. We first found that the Phase 1 duration is controlled by TCR signaling driven by positive selection. In fact, low-affinity HY-TCR-dependent signaling was found to cease quickly(after approximately 19 hours) because the interaction between the low-affinity TCR and the MHC-I molecule is highly dependent on the CD8 molecule; thus, a relatively small decrease in the surface CD8 expression(IM1 in Fig.3) results in the disruption of the TCR-MHC complex. In contrast, the duration of high-affinity OTI-TCR-dependent signaling is long(approximately 53 hours); it lasts until the surface CD8 expression decreases to a level that is low enough to disrupt TCR signaling(IM2 in Fig.3). Interestingly, however, we found that low-affinity HYTCR requires a long phase 2 period to become CD8 T cells (approximately 79 hours); whereas, highaffinity OTI-TCR has a short phase 2(approximately 33 hours). As a result, the total time for positive selection is similar for both T cells[2]. How can this phenomenon be explained? Delicate experiments revealed the crucial relationship between phases 1 and 2, in which the duration of phase 1 determines the cytokine responsiveness of IM cells so that the duration of phase 2 is influenced by the duration of phase 1. For instance, a longer phase 1 duration with high-affinity TCRs such as OTI-TCR increases the cytokine(such as IL-7) responsiveness of IM cells so that the duration of IL-7-driven phase 2 becomes shorter. In contrast, the IL-7; thus, phase 2 takes longer(Fig.3). In this way, immature developing T cells that express various TCRs, each of which have a different affinity to self-MHC-I, can efficiently differentiate into mature CD8 T cells within a certain time[2].
The fact that the CD4/CD8 lineage choice is determined by the duration of TCR signaling made us wonder whether an extremely-high-affinity MHCI- restricted TCR would result in them erroneously becoming CD4 T cells. To address this question, we utilized CD8.4 knock-in mice, in which the CD8.4 gene is knocked into the CD8 α gene locus[9]. The CD8.4 molecule is a chimeric molecule that consists of the extracellular domain of CD8α gene and the intracellular domain of the CD4 gene; thus, CD8.4-dependent MHC-I signaling becomes stronger than CD8-dependent MHC-I signaling, although the interaction between CD8.4-MHC-I and CD8-MHC-I is the same[9]. We introduced the CD8.4 allele into MHC-I restricted TCR Tg mice, and found that low-affinity TCRs such as HY-TCR or F5-TCR with the CD8.4 allele result in more positively selected cells; however, the erroneous development of CD4 T cells was not observed[2,9]. In contrast, high-affinity TCRs such as OTI-TCR with the CD8.4 allele resulted in lineage errors, in which some OTI T cells with the CD8.4 molecule developed into CD4 T cells, even though OTI TCR is an MHC-Irestricted TCR. Accordingly, we revealed that“ errorfree MHC-I positive selection requires phase 1 to end within 53 hours”[2]. In general, the cytosolic tail of the CD8 molecule produces weaker signals than the CD4 molecule; this aspect, which is designed by Mother Nature, must be important for error-free CD8 T cell development.
Studies in recent decades have revealed a great deal of information about the molecular mechanism of T cell development in the thymus; however, new knowledge always open the door to new questions. The thymus produces all types of T cells, not only regular CD4+ and CD8+ T cells, but also innate T lymphocytes, such as natural killer T cells and mucosal-associated invariant T cells. These innate lymphocytes have regulatory functions and contribute to immune homeostasis and immune responses; however, the molecular mechanism of innate T lymphocyte development is still not fully understood. It is important to understand how these T cells recognize self-antigens(or nonself-antigens) in the thymus and how that recognition determines their lineage choice. The elucidation of the detailed mechanisms of thymocyte development is important for understanding the body’s immune system.
I thank Professor Toshinori Nakayama in the Department of Immunology, Graduate School of Medicine at Chiba University for his great support and for his critical reading of this manuscript. I deeply appreciate the continuous help and suggestions from Dr. Alfred Singer in the Experimental Immunology Branch at the National Cancer Institute/National Institutes of Health in the United States. I would also like to thank The Chiba Medical Society for giving me The 9th Chiba Medical Society Award.
Address correspondence to Dr. Motoko Y. Kimura.
Department of Medical Immunology and Department of Immunology Graduate School of Medicine, Chiba University,
1-8-1, Inohana, Chuou-ku, Chiba 260-8670, Japan.
Phone: +81-43-226-2966. Fax: +81-43-226-2966.
E-mail:kimuramo@chiba-u.jp
