




Chiba Medical J. 97E:11-16, 2021
doi:10.20776/S03035476-97E-1-P11
[ Chiba Medical Society Young Investigator Award Minireview ]
Yukari Takeno1,2) , Kazuko Kita2) , and Atsushi Kaneda2)
1) School of Medicine, Chiba University, Chiba 260-8670.
2) Department of Molecular Oncology, Graduate School of Medicine, Chiba University, Chiba 260-8670.
(Received October 16, 2020, Accepted November 5, 2020, Published February 10, 2021.)
Cancer originates via cumulative genetic and epigenetic alterations; it is therefore categorized into molecular subtypes by employing techniques such as microarray analysis and next-generation sequencing, which process conclusive genomic and epigenomic information. Such strategies would aid in classifying patients into different groups that can be subjected to distinct molecular target therapies, and to discover novel cancer driver genes and tumor evolution mechanisms, in addition to identifying underlying drug resistance pathways. Recent development of many molecular targeted therapies has resulted in successful clinical treatment of various cancers. Epigenetic anomalies are potentially reversible; hence, reprogramming the epigenetic profile of cancer cells is a promising strategy for devising innovative therapeutic methods. Herein, we review cancer stratification in the context of molecular subtypes and targeted therapy development.
Cancer, molecular targeted drug, monoclonal antibody, small-molecule inhibitor, epigenetic inhibitor
Cancer is a leading cause of mortality in Japan, despite significant advances in cancer pathogenesis and treatment thus far. Although progress has been achieved in the preliminary detection and surgical excision of colon cancer malignancies in the initial phase, there is minimal change in the mortality rate due to advanced colorectal tumors[1]. Once cancer cells have invaded and metastasized, treatment to eliminate malignancies completely by surgery, chemotherapy, or radiation could prove ineffectual, and an alternate therapeutic approach may be necessary for the recurrent tumor. Cancer is believed to be initiated via the accumulation of genetic and epigenetic modifications, and is therefore divided into certain molecular subtypes using comprehensive genomic and epigenomic inputs[2]. It is necessary to elucidate the detailed molecular mechanisms of tumorigenesis for each subtype and develop specific therapeutic strategies targeting the uncovered driver signaling of the subtype. In the present mini-review, stratified medicine for cancer and currently available targeted therapies have been described.
In the conventional grading system, most cancers have been categorized in terms of their tissues of origin, their stages of clinical progression, and their histopathological features. This traditional categorization, however, mixes up various cancer subtypes with their individual molecular profiles, treatment responses, and clinical prognoses, which may produce unsuccessful results in the applied cancer therapeutic procedure. Anti-cancer treatment regimens are known to generate multiple adverse side effects, and they may even lead to the development of second-site cancers. In the early 1980s, for instance, cyclophosphamide was administered to breast cancer patients at the standard dose and intensified the risk for subsequent development of acute myelogenous leukemia (AML) by several fold. This was due to themutagenic impact of the drug; however, the incidence of second-site cancers has significantly reduced following modern treatment methods that entail lower doses of the drug. These findings indicate the need for more evolved diagnostic tools that can accurately predict responsiveness to various anti-cancer treatments and avoid the use of unnecessary and ineffective therapies. In addition, because the current generation of anti-cancer drugs has been developed based on driver mechanisms of cancer initiation and progression, cancer stratification through detailed molecular assessment is required for effective clinical management[3-5]
Recent technologies for genomic analyses such as microarray and next-generation sequencing have facilitated a comprehensive analysis of molecular aberrations in cancer including chromosomal defects, such as inversion and deletion, as well as copy number changes due to missing or duplicated genomic regions[6,7]. Using unsupervised hierarchical clustering analysis or other stratification methods, cancer cases are classified into several molecular subtypes with distinct aberrations in chromosomal levels, genomic sequence, epigenomic status, or gene expression, which often correlate with specific clinicopathological factors including prognosis, age, bacterial or viral infection, tumor location, smoking, and other tumor risk factors. Such strategies would enable classifying patients into groups undergoing distinct cancer treatment, including targeted therapies, and also accelerate the investigation of tumor evolution and identification of novel cancer driver genes and resistance mechanisms against cancer therapies.
Breast cancer classification, for example, has gradually shifted from a morphologic-based approach to a more integrated methodology, considering clinical characteristics and development of biomarkers[8]. The estrogen receptor (ER) and/or the progesterone receptor (PgR) , expressed in more than 75% of breast cancers, possess predictive and prognostic value as biomarkers[8,9]. In addition, the receptor protein HER2/ERBB2 is expressed in approximately 10-15% of breast cancers. HER2, a receptor tyrosine kinase involved in cell growth regulation, is also considered a prognostic marker due to the emergence of HER2- targeted therapies[10]. These biomarkers are often identified using immunostaining and in situ DNA hybridization techniques concordant with international guidelines[8]. Over the last few decades, abundant breast cancer specimens have undergone comprehensive genomic and molecular studies. The major cohorts, such as The Cancer Genome Atlas (TCGA) project, the International Cancer Genome Consortium (ICGC) , and the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) , in which high-resolution comprehensive studies have been conducted at various levels, aimed at improving the existing grading scheme by integrating extensive molecular profiling, and helped to identify breast cancer cases that may profit by more innovative therapies[8]
Two thoroughly evaluated and substantiated molecular classifiers have been developed to date: PAM50/intrinsic subtypes and integrative clusters. In 2000, the intrinsic subtypes emerged through analysis of gene expressions in breast cancer specimens using cDNA microarrays as follows[11]. From approximately 9,000 genes analyzed, a total of ~550 genes were extracted as the intrinsic gene subset, exhibiting the phenotype of individual tumors; their expression profiles varied to a higher degree between distinct types of tumors than between paired samples from the same tumors. Hierarchical clustering analysis based on the gene-expression patterns revealed clusters of samples termed“ intrinsic subtypes”[12]. In 2013, US FDA approved a prediction analysis of the microarray 50 gene set (PAM50) , which was modified from the original intrinsic subtype definition, as a diagnostic assay. PAM50 analysis allocated each breast cancer sample to the luminal A, luminal B, HER2-enriched, and basallike subtype, based on the ER and HER2 expression patterns[13]. The stratification by PAM50 shows a different prognosis especially in patients without treatment, when stratified by ER status. In addition, a numeric score predicting the risk of recurrence will be provided by PAM50 and can extract patients who may profit by adjuvant treatment from ER-positive patients[13,14]. While the above subtypes defined by gene expression analyses were provided, tumor subtypes based on similarity of copy number alterations (CNAs) were discovered[8,15]. Driver genes elucidated through combination of gene expressions and CNAs classify breast tumors into ten novel subtypes termed“ integrative clusters”, with distinct clinical outcomes[8,16]
Recently, several molecular targeted therapies have been developed and approved, resulting in the successful clinical treatment of various cancers including breast, leukemia, colorectal, lung, and ovarian cancers[17,18]. For example, trastuzumab, a humanized IgG1 monoclonal antibody, can be chosen to treat breast cancer patients with overexpression of the oncogene HER2. The HER2-positive status portends a poor diagnosis but it also predicts response to trastuzumab[18]. A total of 11,991 breast cancer patients were enrolled in eight randomized controlled trials, resulting in a reduction in cancer mortality by one-third when HER2 -overexpressing breast cancer patients were administered trastuzumab in addition to the standard chemotherapy regimens. This cohort also showed a 40% reduction in the recurrence rate[19]. In addition to monoclonal antibody therapies including trastuzumab (Table 1) , small-molecule inhibitors against multiple driver kinases have been developed (Table 2).
Research over the past few decades has revealed that epigenetic aberrations are central to the cancer initiation and progression[2,20].“ Epigenetics” is the analysis of heritable changes in regulation of gene expression that are caused by modification of genomic DNA, not by changes in DNA sequence itself. The epigenome is composed of specific modifications in components of chromatin, primarily genomic DNA and histones. Epigenetic modifications are deposited by enzymes termed“ writers”, subsequently recognized by effector proteins (“readers”) , and removed by specific enzymes (“erasers”) [21]. These epigenetic mechanisms regulate chromatin structure and provide a differential gene expression program without modifying the genomic DNA sequence. Unlike genetic mutations, which are presumably impossible to reverse, epigenetic aberrations are potentially reversible. Therefore, the ability to reprogram the epigenetic landscape in the cancer epigenome and restoration of a“ normal epigenome” is one of the most promising targets for novel therapies[20,22]. In addition, the factors modifying the epigenome, including readers, writers, and erasers, are enzymes that recognize specific covalent modifications and can therefore serve as ideal targets for smallmolecule drugs[21]
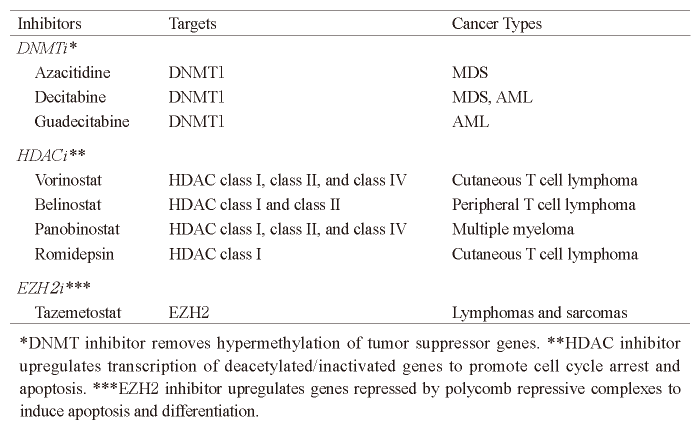
Currently, reagents that effectively reverse the aberrations in DNA methylation and modification of histone tails occurring in tumor have been approved or have entered clinical trials (Table 3) [21]. DNA methylation is known as a stable gene-silencing mechanism that plays a crucial role in modulating gene expression and chromatin structure[20]. Two nucleoside analogs with inhibitory activity against DNA methyltransferase, 5-azacytidine (azacitidine) and 5-aza-2’-deoxycytidine (decitabine) , reduce DNA methylation levels, activate the expression of genes such as tumor-suppressor genes aberrantly repressed in cancer, and induce apoptosis or growth arrest in cancer cells. In clinical trials of these inhibitors, more than 15% of patients with myelodysplastic syndrome (MDS) or AML favorably responded to the epigenetic treatment, as demonstrated by a decrease in malignant cell burden and prolonged survival, so that FDA approved these inhibitors for clinical treatment[23,24]. A secondgeneration reagent to reduce DNA methylation levels, guadecitabine, enhanced pharmacology and pharmacodynamic effects, and has shown promising results in early clinical trials.
A concomitant loss of histone acetylation is also linked to aberrant gene repression in cancer. Restoration of patterns of normal histone acetylation via treatment with histone deacetylase (HDAC) inhibitors has been found to exert anti-cancer effects, which are related to the reactivation of silenced tumor-suppressor genes[21]. FDA has approved vorinostat, belinostat, and romidepsin for the treatment of cutaneous or peripheral T-cell lymphomas. In addition, FDA recently approved panobinostat for the treatment of drug-resistant multiple myeloma in the case of combination use with bortezomib, a proteasome inhibitor.
These drugs have been developed to target molecules that show specific aberrations in cancer, for example, mutation, activation, or inactivation. However, molecules that maintain normal functions in cancer cells could be targeted and utilized for stratified therapeutic strategies. The research conducted by Takeno, which received the 2020 Encouraging Prize from the Chiba Medical Society, is an analysis of a compound that shows antiproliferative activity specifically against cancer cell lines with wild-type p53. The compound exerts its anti-cancer effect via the normal function of p53, and is therefore ineffectual against cancer cells harboring the p53 mutation (unpublished data).Utilization of specific molecular features in each subtype, including normal functions, may serve as another strategy to develop novel therapeutics.
Table. 1 Monoclonal antibody therapy targeted toward specific cancer subtypes

Table. 2 Small-molecule inhibitors
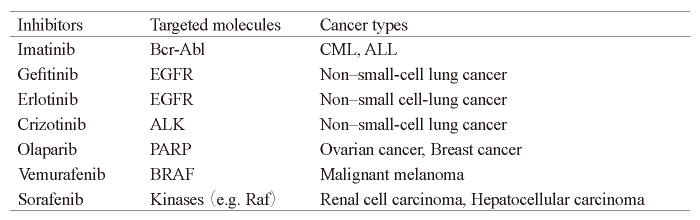
Table 3 Epigenetic inhibitors Inhibitors Targets Cancer
Stratification of cancer based on comprehensive analyses helps in unraveling the detailed mechanisms of tumorigenesis. Further research on critical aberrations in each molecular subtype of cancer is essential and is anticipated to lead to the development of additional molecular targeted therapies to impart clinical success in cancer treatment.
Y.T. wrote the original draft of the paper. K.K. reviewed and edited the paper. A.K. conceptualized, reviewed and edited the paper. All authors approved the final version of the manuscript.
Therapeutics Research Initiative Grant from Chiba University School of Medicine 2019-S3 (Y.T.) , and Global and Prominent Research grant 2018-Y9 from Chiba University (A.K).
A.K. is a member of the Editorial Board of the Chiba Medical Journal. The other authors declare that they have no conflict of interest to disclose.
Not applicable.
Not applicable.
The authors thank all the university members who supported their study, especially Prof. Tetsuhiro Nemoto for providing candidate compounds.
Address correspondence to Dr. Atsushi Kaneda.
Department of Molecular Oncology, Graduate School of
Medicine, Chiba University, 1-8-1, Inohana, Chuou-ku, Chiba
260-8670, Japan.
Phone/Fax: +81-43-226-2039.
E-mail:kaneda@chiba-u.jp
