




Chiba Medical J. 97E:17-24, 2021
doi:10.20776/S03035476-97E-2-P17
[ Chiba Medical Society Award Review ]
Akira Suto1,2)
1) Department of Allergy and Clinical Immunology, Graduate School of Medicine, Chiba 260-8670.
2) Institute for Global Prominent Research, Chiba University, Chiba 260-8670.
(Received November 14, 2020, Accepted December 9, 2020, Published April 10, 2021.)
IL-21 is produced by Th17 cells and follicular helper T cells. It is an autocrine growth factor and plays critical roles in autoimmune diseases. In autoimmune mouse models, excessive production of IL-21 is associated with the development of lupus-like pathology in Sanroque mice, diabetes in NOD mice, autoimmune lung inflammation in Foxp3-mutant scurfy mice, arthritis in collagen-induced arthritis, and muscle inflammation in experimental autoimmune myositis. IL-21 production is induced by the transcription factor c-Maf following Stat3 activation on stimulation with IL-6. Additionally, Stat3 signaling induces the transcription factor Sox5 that along with c-Maf directly activates the promoter of RORγt, a master regulator of Th17 cells. Moreover, T cell-specific Sox5-deficient mice exhibit decreased Th17 cell differentiation and resistance to experimental autoimmune encephalomyelitis and delayedtype hypersensitivity. Another Sox family gene, Sox12 , is expressed in regulatory T cells (Treg) in dextran sulfate sodium-induced colitic mice. T cell receptor-NFAT signaling induces Sox12 expression that further promotes Foxp3 expression in CD4+ T cells. In vivo, Sox12 is involved in the development of peripherally induced Treg cells under inflammatory conditions in an adoptive transfer colitis model. This review highlights the crucial roles of transcription factors in the onset of autoimmune diseases and the differentiation of IL-21-producing CD4+ T cells, Th17 cells, and Treg cells.
autoimmune disease, IL-21, c-Maf, Sox5, Sox12
Autoimmune diseases are caused by the breakdown of tolerance to self-antigens and are characterized by the activation of T cells, including pathogenic IL- 17-producing helper T cells (Th17 cells) and IL-21- producing CD4+ T cells. Th17 cells are eventually differentiated in the presence of IL-23 and IL-21, and further produce IL-17A, IL-17F, and IL-21. On the other hand, regulatory T (Treg) cells, defined by the expression of Foxp3, play a central role in protecting against excessive inflammatory responses caused by autoimmune diseases. This review highlights the crucial roles of transcription factors in the onset of autoimmune diseases and the transcriptional regulation of the differentiation of Th17 cells, IL-21-producing CD4+ T cells, and Treg cells.
IL-21 is a four-helix-bundle type I cytokine with significant homology to IL-2, IL-4, and IL-15 [1]. In vivo, IL-21 has been shown to be involved in many kinds of autoimmune disease models. For example, lymphopenia induced IL-21-mediated homeostatic expansion has been shown to develop type I diabetes in NOD mice [2,3]. Excessive production of IL- 21 is associated with high titers of auto-antibodies and development of lupus in Sanroque mice [4]. Furthermore, neutralization of IL-21 by IL-21-receptor (IL-21R) Fc chimera protein has been reported to ameliorate conditions in mouse models of lupus and rheumatoid arthritis [5,6].
Mutations in Foxp3 gene lead to development of IPEX (Immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome in humans [7]. Scurfy mice have a frame-shift mutation in Foxp3 gene. They completely lack Treg cells and suffer from autoimmune diseases, such as skin, lung, and liver inflammation, leading to death within 4 weeks of birth [8]. Activation of CD4+ T cells caused by the lack of Treg cells plays a crucial role in the onset of autoimmune diseases in scurfy mice [9]. Among the subset of activated CD4+ T cells, unique IL-21-producing c-Maf+ CD4+ T cells develop, which further induce short-lived effector CD8+ T cells (SLEC) and accelerate multi-organ autoimmune inflammation in scurfy mice [10]. IL-21 receptor deficiency results in reduced multi-organ inflammation, prolonged survival, and decrease of SLEC in the lung in scurfy mice. Thus, IL-21-producing CD4+ T cells are extensively involved in the onset of autoimmune lung inflammation in scurfy mice, presumably through the induction of SLEC. Further, Sharma et al. have shown that intramuscular injection of lymph node (LN) cells from scurfy mice into recombination activating gene (RAG) -deficient mice causes inflammatory myopathy (IM) -like pathological changes in uninjected muscles [11,12]. This suggests the possible involvement of IL-21 in the pathogenesis of IM. Polymyositis (PM) and dermatomyositis (DM) are the major subgroups of idiopathic IM characterized by infiltration of inflammatory cells into the affected muscles [13]. Regarding the relationship between IL-21 and IM in humans, CXCR3- CXCR5+ helper T cells, which produce IL-21, increase in peripheral blood in juvenile DM patients having skin rash and muscular weakness [14]. In addition, serum levels of IL-21 are increased in the subset of patients with IM as compared to those in healthy controls [15].
In experimental autoimmune myositis (EAM) IM mouse model, the severity of EAM is diminished in IL-21-/- mice [15]. GM-CSF production from γδT cells, but not Th17 cell differentiation, is significantly reduced in EAM-induced IL-21-/- mice. Importantly, the neutralization of GM-CSF or deficiency of γδT cells significantly improves muscle weakness and muscle inflammation in EAM-induced mice. Major GM-CSF producers in EAM-induced mice muscles are Vγ4+ Vδ4+ CD27- cells that are significantly decreased in EAM-induced IL-21-/- mice. Intriguingly, Vγ4+ Vδ4+ CD27- cells express high levels of CX3CR1. In addition, CX3CL1, a ligand of CX3CR1, is induced in the muscle upon EAM induction in WT mice but not in IL-21-/- mice. In summary, IL-21 facilitates autoimmune myositis through the accumulation of GMCSF- producing Vγ4+Vδ4+ cells in the muscle possibly via the CX3CR1-CX3CL1 pathway.
Activated CD4+ T cells differentiate into at least three distinct helper T cell subsets as defined by their patterns of cytokine production. Th1 cells produce IFN-γ and lymphotoxin and protect against intracellular pathogenic infections. Th2 cells produce IL-4, IL- 5, and IL-13 and are essential for defending against parasites. On the other hand, Th17 cells produce IL- 17A and IL-17F and play a pathogenic role in a variety of autoimmune diseases. IL-21-producing CD4+ T cells develop preferentially in Th17-polarizing condition (IL-6+TGF-β+anti-IL-4 antibody+anti-IFN-γ antibody) . Among these, IL-6+anti-IL-4 antibody+ anti-IFN-γ antibody strongly induce the development of IL-21-producing CD4+ T cells without inducing the production of IFN-γ, IL-4, IL-5, IL-13, IL-17A, and IL-17F [16]. While TGF-β inhibits IL-6-mediated differentiation of IL-21-producing CD4+ T cells in a dose-dependent manner, IL-21 itself induces the differentiation of IL-21-producing CD4+ T cells.
NFAT directly binds to the IL-21 promoter and activates its transcription in Th2 cells [17]. On the other hand, T-bet suppresses IL-21 transcription by inhibiting the binding of NFAT to the promoter in Th1 cells. IRF-4 binds to and activates IL-21 promoter; whereas, IRF-4-binding protein (IBP) inhibits IL-21 production by regulating the activity of IRF-4 [18]. Stat3 (activated by IL-6 and/or IL-21) is required for IL-21 production in Th17 cells [19]. However, RORγt, the master regulator of Th17 cells, is not involved in the production of IL-21 [19]. To determine the transcription factor that induces IL-21-producing CD4+ T cells, DNA microarray analysis of IL-6-stimulated CD4+ T cells was performed. Expression of c-Maf, a basic leucine zipper protein, is significantly upregulated in IL-21- producing CD4+ T cells [20]. Overexpression of c-Maf strongly induces IL-21-producing CD4+ T cells without IL-6 stimulation or autocrine effect of IL-21. c-Maf directly binds to and activates both IL-21 promoter and CNS-2 enhancer through Maf recognition elements (MAREs) , TGCn6-8GCA (Fig. 1) . On the other hand, TGF-β upregulates IL-6-induced c-Maf expression but inhibits c-Maf-induced IL-21 production in CD4+ T cells. Additionally, Foxp3 binds to IL-21 promoter and CNS-2 enhancer and inhibits c-Maf-induced IL-21 production in CD4+ T cells. In summary, c-Maf directly induces IL-21 production by activating IL-21 promoter and CNS-2 enhancer and TGF-β suppresses c-Mafmediated IL-21 production in CD4+ T cells, presumably through Foxp3.
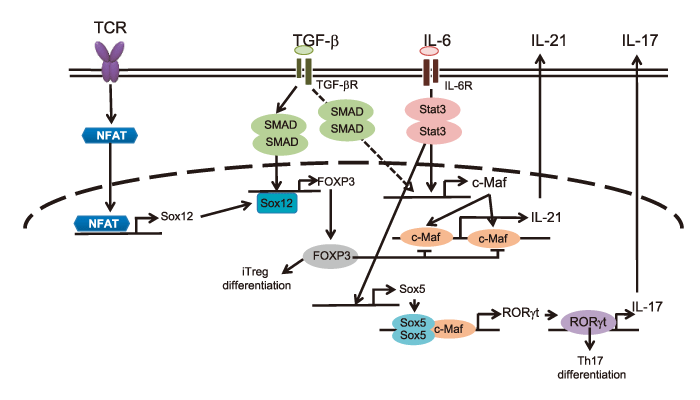
Fig. 1 Roles of Sox5 and c-Maf in the production of IL-17A and IL-21. Under Th17-differentiating conditions, IL-6 induces Sox5 expression in CD4+ T cells in a Stat3-dependent manner. Additionally, IL-6 induces c-Maf expression especially in the presence of TGF-β. Sox5 and c-Maf co-induce IL-17A production via induction of RORγt. c-Maf induces IL-21 by binding to the IL-21 promoter and CNS-2 enhancer. On the other hand, TGF-β inhibits c-Maf-induced IL-21 production in CD4+ T cells. TGF-β-induced Foxp3 binds to IL-21 promoter and CNS-2 enhancer and inhibits c-Maf-induced IL-21 production in CD4+ T cells.
Th17 cells produce IL-17A and IL-17F and play a pathogenic role in many autoimmune diseases. Since ectopic expression of RORγt (encoded by Rorc) induces differentiation of Th17 cells while RORγt-deficient mice lack Th17 cell differentiation [21], RORγt is regarded a master regulator of Th17 cells [22]. Additionally, IL- 6- and/or IL-21-mediated activation of Stat3 plays a central role in RORγt-induced development of Th17 cells [23-26]. Stat3 binds to intron 1 of Rorc gene and promotes trimethylation of histone H3 lysine 4 on Rorc, though Stat3 does not activate Rorc promoter [27,28]. Regarding the downstream pathways of Stat3 signaling, a number of genes including Rora, Maf, Ahr, Irf4 , Batf, Nfkbiz, and HIF-1 α are activated by Stat3 and involved in Th17 cell differentiation [28,29-34]. Among these transcription factors, HIF-1 activates Rorc promoter [34]
Sox5 belongs to the Sox (SRY-related high-mobilitygroup [HMG]-box) family of transcription factors. Sox5 is a member of SoxD family that is composed ofSox6, Sox13, and Sox5 [35,36]. SoxD protein has an HMG domain, which mediates the binding to DNA, and two coiled-coil domains, where the first coiled-coil domain induces dimerization of SoxD proteins. SoxD proteins do not have transrepression or transactivation domains and their activity is probably influenced by the molecules they interact with. Sox5-deficient mice die after birth due to a small thoracic cage and cleft secondary palate; this is consistent with the finding that Sox5 is strongly expressed in chondrocytes, neurons, spermatids, and oligodendrocytes. Regarding the relationship between autoimmune diseases and Sox5, it has been recently reported that Sox5 is one of the most significantly upregulated genes in the blood of patients with multiple sclerosis [37]. In addition, DNA microarray analysis showed that Sox5 gene is the most strongly expressed transcription factor in CD4+ T cells upon IL-6 stimulation. Particularly a novel isoform of Sox5, Sox5t, is expressed in Th17 cells and IL-21- producing CD4+ T cells [38]. Upon stimulation with IL-6, Stat3 induces the expression of c-Maf and Sox5t. Subsequently, c-Maf along with Sox5t induces the expression of RORγt by directly activating the RORγt promoter, suggesting that Sox5t functions downstream of IL-6-Stat3 signaling and upstream of RORγt expression during the differentiation of Th17 cells (Figure. 1)
Additionally, Sox5 plays a crucial role in the onset of Th17 cell-mediated autoimmune models. Experimental autoimmune encephalomyelitis (EAE) is a mouse model of multiple sclerosis mainly caused by Th17 cellmediated autoimmune responses. In this model, CD4cre Sox5fl/fl mice exhibit significantly reduced inflammatory cell infiltration and clinical scores as compared to the control Sox5fl/fl mice. Further, the number of Th17 cells in the brain and spinal cord decreases in CD4cre Sox5fl/fl mice. In addition, the severity of delayed-type hypersensitivity (DTH) reduces in CD4cre Sox5fl/fl mice as compared with that in the control Sox5fl/fl mice. Furthermore, mice injected with c-Maf- and Sox5texpressing CD4+ T cells exhibit strong DTH response and IL-17 production in the draining LNs. Collectively, Sox5 is crucially involved in the development of Th17 cell-mediated inflammatory responses.
Treg cells are essential for maintaining immune tolerance in the gut where food antigens and microbiota are present. Among Treg cells, thymus-derived Treg (tTreg) cells are indispensable for maintaining immune tolerance to self-antigens and peripherally induced Treg (pTreg) cells play essential roles in the composition of commensal microbiota and repression of allergic inflammation in mucosa [39]. pTreg cells arise from expression of Foxp3 during CD4+ T cell differentiation in the periphery and consist of a majority of gut Treg population [40]. In autoimmune colitis models, pTreg cells together with tTreg cells act to restore immune tolerance [41]. These findings suggest that pTreg cells are crucial in suppressing gut inflammation.
Regarding the underlying mechanism of Foxp3 induction during T cell differentiation in the periphery, strong T cell receptor (TCR) stimulation with suboptimal co-stimulation, IL-2, TGF-β, microbial metabolites, and retinoic acid induce pTreg cell differentiation both in vitro and in vivo [42-45].
Under inflammatory conditions, Treg cells enhance suppressive activity, acquire an activated phenotype, and increase their population [46,47]. Regarding the underlying mechanism of maintaining activated Treg cells, Arvey et al. have shown that Foxp3 modulates the expression of its target genes by inducing repressive histone H3 marks under inflammatory conditions [48]. This study has uncovered the importance of Foxp3- mediated transcriptional regulation in activated Treg cells under inflammatory conditions.
RNA sequencing analysis of Treg cells in dextran sulfate sodium-induced colitic mice reveals that Sex determining region Y box 12 (Sox12) is the only transcription factor that is significantly induced in Treg cells [49]. Sox12 is a member of the SoxC family that is composed of Sox4, Sox11, and Sox12. SoxC proteins have a C-terminal transactivation domain and an N-terminal HMG box domain that mediates binding to DNA. These proteins play crucial roles in the development of the nerve system, pancreas, and kidneys [50]. Regarding immunological aspects, Sox4 is involved in survival of the B cell precursor and differentiation of Th2 cells [51]. Although Sox12 and Sox4 but not Sox11 are expressed in Treg cells, TCR stimulation strongly upregulates Sox12 expression but downregulates Sox4 expression in Treg cells. Upon TCR stimulation, NFAT is activated and it binds upstream of exon 1 of Sox12 gene locus. Thus, TCR-NFAT signaling is important for inducing Sox12 expression in CD4+ T cells [49]. In addition, Sox12 is involved in the differentiation of pTreg cells under inflammatory conditions in colitic mice, though Sox12 is not essential for the development of tTreg cells. Moreover, overexpression of Sox12 is sufficient to induce Foxp3 expression in CD4+ T cells even in the absence of IL-2 or TGF-β where Sox12 binds to the Foxp3 promoter and drives its transcription (Figure. 1) . In summary, TCRNFAT signaling induces the development of pTreg cells in colitic mice partly through Sox12 induction.
Recent studies have revealed how Sox family proteins regulate the development of Th17 cells, Treg cells, as well as the onset of autoimmune diseases. Sox5, a protein of SoxD family, induces Th17 cell differentiation. Sox12, a protein of SoxC family, is required for pTreg differentiation. However, the role of Sox4, another protein of the SoxC family in the regulation of Treg cells is still largely unknown. Thus, further research on elucidating the mechanism underlying development of Treg cells is needed.
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, the Japanese Government, and LGS (Leading Graduate School at Chiba University) Program, MEXT, and Institute for Global Prominent Research, Chiba University, Japan.
The author declares no conflict of interest associated with this manuscript.
Not applicable.
Not applicable.
The author expresses gratitude to Prof. Hiroshi Nakajima for critically reading the manuscript.
Address correspondence to Dr. Akira Suto.
Department of Allergy and Clinical Immunology, Graduate
School of Medicine, Chiba University, 1-8-1 Inohana, Chiba 260-8670, Japan.
Phone: +81-43-226-2198. Fax: + 81-43-226-2199
E-mail:suaki@faculty.chiba-u.jp
