




Chiba Medical J. 98E:1-7, 2022
doi:10.20776/S03035476-98E-1-P1
〔 Chiba Medical Society Young Investigator Award Minireview 〕
Takahiro Fujii1,2), Atsushi Okabe2), and Atsushi Kaneda2)
1) School of Medicine, Chiba University, Chiba 260-8670.
2) Department of Molecular Oncology, Graduate School of Medicine, Chiba University, Chiba 260-8670.
(Received October 15, 2021, Accepted October 28, 2021, Published February 10, 2022.)
Epstein-Barr virus (EBV) is known as an oncogenic virus and causes tumors in a variety of tissues, including lymphocytes and epithelial cells. EBV infects host cells latently as an episome and affects the host genome and epigenome while switching between three types of latent stages and a lytic stage. In recent years, the development of analytic methods using next generation sequencing and chromosome conformation capture technologies has enabled us to identify abnormalities at the molecular level in cancer cells. In EBV-positive tumors, epigenetic alterations such as DNA methylation, viral miRNA-mediated expression changes, and aberrations in histone modification and 3D chromatin structure, together with the involvement of EBV, have been revealed by analysis of tumor cells and in vitro EBV infection experiments. Further understanding of host-EBV interactions and their roles in the cancer pathogenesis is expected to lead to the development of revolutionary therapies targeting the epigenome. Here we review epigenetic contribution to tumorigenesis by EBV infection in host cells.
Epstein-Barr virus (EBV) , epigenome, DNA methylation, chromatin structure
EBV is the first discovered human oncovirus, isolated from Burkitt lymphoma (BL) cells; it is a gamma-herpes virus with 184 kb of double-stranded DNA. It is estimated that about 95% of the world’s population is latently infected with EBV in memory B cells, causing more than 200,000 cancers each year. EBV is a major cause of B cell tumors such as BL, diffuse large B cell lymphoma (DLBCL) , and Hodgkin lymphoma, as well as extranodal natural killer/T cell lymphoma (ENKTL) , chronic active EBV infection (CAEBV) , gastric cancers (GC) , and nasopharyngeal carcinoma (NPC) (Table 1) [1]. There are three different latency types depending on the host cells, and the viral gene expression is different depending on the type.The mechanisms of EBV-induced carcinogenesis include DNA methylation, viral miRNAs, histone modifications and chromatin structural abnormalities (Table 2) . In this mini-review, we will review these epigenetic aberrations and their contribution to tumorigenesis.
Table 1 EBV-associated tumors with different types of viral latency
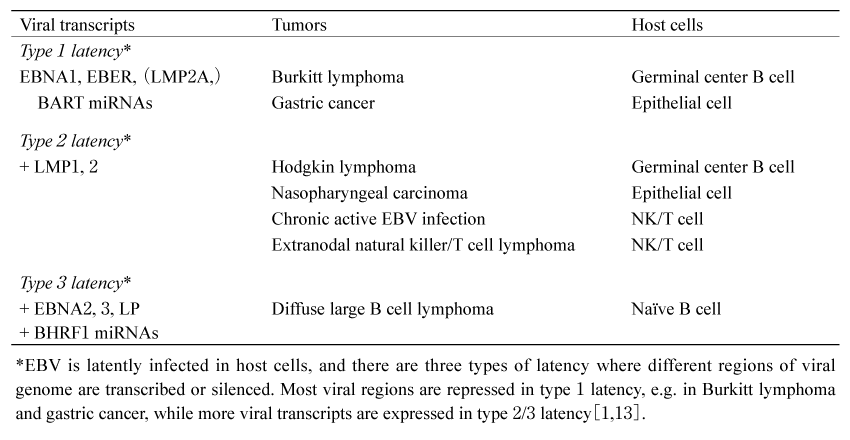
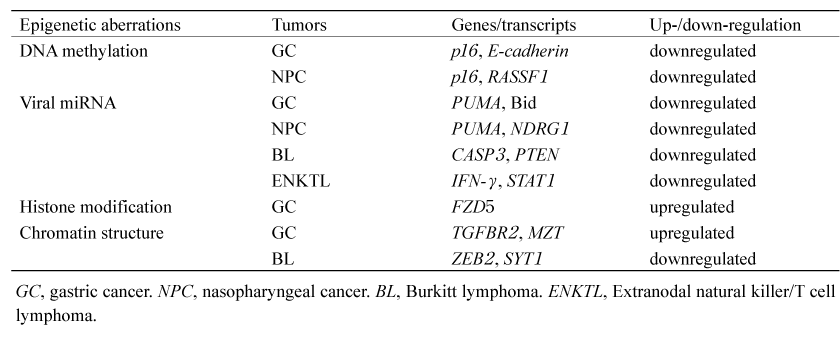
Table 2 Reported epigenetic alterations in EBV-associated tumors[5,10,11,19,31,32]
DNA methylation is a reaction in which a methyl group binds to cytosine, and in human genomic DNA, it occurs mainly in cytosine-guanosine dinucleotides (CpGs) and has the effect of inhibiting gene transcription. Highly enriched regions of CpGs, called CpG islands, are found around promoter regions of genes, and DNA methylation can inactivate the expression of any potentially harmful viral sequences or transposons in heterochromatin, or play an important role in genomic imprinting and X-chromosome inactivation in women. However, promoter DNA hypermethylation of tumor suppressor genes and genes related to cell adhesion can inactivate their expression and lead to cancer development. Gene silencing by promoter hypermethylation has been reported in a variety of cancers. The evaluation of genomic DNA methylation in cancer cells is very important because these genes are therapeutic targets for methylation inhibitors [2].
Latent Membrane Proteins (LMP) 1 and 2A expressed by EBV are known to induce cellular DNA methyltransferases and affect the methylation status of the host genome in EBV-infected cells[3]. A comprehensive DNA methylome analysis using clinical gastric cancer samples reported that gastric cancer can be classified into three epigenotypes: low- and highmethylation epigenotypes in EBV (-) cancers and EBV (+) specific markedly higher methylation epigenotypes. It has been confirmed that hypermethylation is brought about by EBV infection of GC cell lines. The functions of genes with promoter hypermethylation in EBV (+) -epigenotypes include negative regulation of cell communication (HCRT, CAV1, GRB10, DLL3, CXXC4) and perinuclear region of cytoplasm (SLC11 A2, HCRT, CAV1, CABP1) [4].
Promoter hypermethylation is also observed in EBV (+) NPC. Some of the notable genes include p16 and RASSF1. p16 is a growth suppressor with G0/G1 arrest and inhibition of interaction between cdk4/cdk6 and CCND1. RASSF1 is associated with TGF-β signaling and acts as a growth suppressor by inducing activin βE and suppressing Id2, which promotes cell proliferation, oncogenesis and invasion[5]. It also suppresses tumorigenesis by maintaining microtubule stability[6]. Promoter hypermethylation has also been observed in WIF1 and TSLC1, which are tumor suppressor genes in NPC. Other gene pathways in which promoter hypermethylation is observed include cell invasion and migration, e.g., CDH1 and ZMYND10, response to DNA damage, e.g., GADD45, apoptosis, e.g., DAPK1 and UCHL1, cell cycle and proliferation, e.g., DLC1 and RARB, MAPK signaling, e.g., RASAL and DAB2, and Wnt/β-catenin signaling, e.g., SOX1[5].
Pfeffer et al. were the first to isolate five EBV miRNAs from a Burkitt lymphoma cell line[7]. So far, 49 mature miRNAs have been identified to be encoded by EBV, of which 44 are BART miRNAs and 5 are BHRF1 miRNAs[8]. Recently, several studies have shown that BART miRNAs are involved in carcinogenesis of epithelial cells. miR-BART5 downregulates PUMA and miR-BART9, 11, and 12 downregulate Bim, contributing to the apoptosis resistance. Both PUMA and Bim are the proapoptotic proteins of Bcl-2 family members, and their suppression leads to cellular survival. It has been reported that miRBART4- 5p represses Bid by profiling EBV (+) GC[9]. MiR-BART4-5p is also the proapoptotic protein of Bcl-2 family members, and regulation of apoptosis by repression of Bid is important for gastric carcinogenesis [10,11]. EBV infection of NPC cell lines has revealed that the BART miRNA cluster 2 is a causative factor for down-regulation of NDRG1. NDRG1 is highly expressed in epithelial cells, and it has been reported that NDRG1 interacts with LRP6, Wnt receptor, and functions as a tumor suppressor by blocking Wnt signaling. Immunohistochemical analyses have confirmed that NDRG1 protein expression is also significantly low in EBV (+) NPC specimens[12].
In contrast, by whole EBV sequencing, it has been found that the BART miRNA cluster regions are mainly deleted in patients with CAEBV, ENKTL and DLBCL. In particular, miR-BART6-5p and miR-BART6-3p that negatively regulate BZLF1 and BRLF1, are frequently deleted[8]. It has been suggested by in vivo models that increased expression of BZLF1 and BRLF1 contributes to lymphomagenesis by promoting lytic EBV replication[13].
The active promoter region of the human genome is marked by enrichment of H3K4me3 and H3K27ac, and the enhancer region is marked by enrichment of H3K4me1 and H3K27ac. H3K9me3 is enriched in the heterochromatin-associated region, and H3K27me3 is in the polycomb complex-mediated transcriptional repression region[14,15].
It was confirmed that EBV infection of gastric cancer cell lines caused a rearrangement of the active histone modifications of H3K4me1 and H3K27ac and the repressive histone modifications of H3K27me3 and H3K9me3. Loss of repressive marks in promoter regions is frequently replaced by de novo DNA methylation to maintain repression, while loss of repressive marks in enhancer regions leads to enhancer activation by induction of active marks. The repressed regions are associated with tumor suppressor pathways and the active enhancer regions are associated with pathways of cancer hallmarks[16,17]. Our further analysis identified the transcription factor ATF3, which could be upregulated by EBV factors, and reported that it targets genes related to the hallmarks of cancer, such as evading growth suppressors and sustaining proliferative signaling[18]. Furthermore, it was found that EHF, which is upregulated by LMP2A, activates the WNT pathway through enhancer activation and contributes to tumorigenesis[19].
Recently, it has been shown that spatial chromatin dynamics plays a major role in the regulation of gene expression and disease pathogenesis[20]. The threedimensional (3D) structure of genome organization includes enhancer-promoter loops[21], topologically associating domains (TADs) [22], and chromosome compartments[23]. The active enhancer forms a chromatin loop structure with the promoter via mediator and cohesin and is involved in the up-regulation of the target gene[24]. This interaction is constrained within higher-level structures called TADs[25]. TADs are submegabase-scale chromatin interaction domains bounded mainly by the insulator binding protein CTCF. Within the domain, genomic DNAs are spatially close to each other and gene expression is dynamically regulated. TADs are highly conserved among different cell types, but it has been reported that in cancer cells, disruption of the domain leads to the formation of ectopic chromatin loops and activation of proto-oncogene expression[26-28]. Furthermore, multiple TADs form an assembly called the chromosome compartment. It has been predicted that compartment formation is due to interactions with various nuclear molecules such as DNA, ncRNA, histone marks, as well as chromatin regulators[29]. The compartments can be divided into A compartment, where gene expression is active, and B compartment, where gene expression is repressed, depending on their epigenetic marks[23]. Currently, Chromosome conformation capture-based techniques are being used to analyze these 3D chromatin structures[30].
Okabe and Kaneda with their colleague discovered a new oncogenic mechanism called“ enhancer infestation” by host-EBV genome interaction through EBV infection of GC cell lines. EBV interacts with heterochromatin regions characterized by high AT content, low gene density, and overlapping with laminin. In EBVIRs, changes in TADs and B-to-A compartment shifts were observed with decreased repressive histone marks and increased active histone marks. These de novo enhancers formed a loop structure with the neighboring oncogene promoters such as TGFBR2 and MZT1, and contributed to activation of the genes and cell proliferation[31].
Thus, we reported that aberrant enhancer activation by EBV infection contributes to carcinogenesis in GC cell lines, while the role of host-EBV interaction in BL cell lines has been reported differently. Kim et al. reported that EBVIRs in BL cells are enriched for EBNA1, EBF1, and RBP-jK binding sites. They found high levels of both H3K9me3 and AT content in the regions and concluded that EBNA1 binding represses the expression of genes in EBV tethering sites via H3K9me3 associated heterochromatin[32]. Stephanie et al. have shown that EBV localizes to the B compartments during latent infection in BL cell lines, but moves to non-lamin-associated domains regions, which are open chromatin regions, at the lysis stage[33]. Wang et al. have shown that in lymphoblastoid cell lines, EBV interacts with open chromatin regions rich in active enhancer marks, e.g., H3K27ac and H3K4me1, which are associated with the super enhancer complex[34]. Collectively, these reports have left us questions on how EBVIRs are determined, whether the interaction contributes to transcriptional activation or repression, and whether there are differences in function depending on cell types, therefore further studies are required.
In addition to EBV, the human papillomavirus (HPV) and the hepatitis B virus (HBV) are known as oncoviruses. Due to a different mechanism of integration, HPV infection is also reported to affect the host genome and epigenome, and the effect of the viral genome itself on the host epigenome is also important. HPV is known to be responsible for cervical cancers, anogenital cancers, and oropharyngeal cancers. It usually infects latently as an episome, but accidental integration into common fragile sites or transcriptionally active regions can lead to carcinogenesis[35]. The c-myc peripheral region is one of the integration targets of HPV, and in aggressive neuroendocrine cervical cancer, it has been reported that integration causes amplification of the MYC gene, which contributes to cell proliferation[36]. HBV has been studied for its interaction with the host as an episome. HBVs are docked to specific heterochromatin regions on chromosome 19 during the latent stage, but interact with the A compartment of the surrounding chromosomes when reactivated[37]. The contact sites are located on CpG islands upstream of transcriptionally active genes, and have been reported to be particularly associated with genes that have abnormal expression changes before and after infection[38]. In addition, although the human adenovirus serotype 5 has not been reported as an oncovirus, changes in TADs and compartment shifts have been reported after infection[38], supporting the idea that the viral episome influences the host epigenome. The contribution of this effect to carcinogenesis needs to be further elucidated.
EBV infection has been found to cause alterations of the host epigenome such as methylated epigenotypes, enhancer infestation and transcriptional rewiring. Further studies on the dynamics of EBV in cells will help us to understand the oncogenic mechanisms of EBV-positive tumors and discover therapeutic targets.
T.F. wrote the original draft of the manuscript. A.O. reviewed and edited the manuscript. A.K. conceptualized, reviewed and edited the manuscript. All the authors read and approved the final version of the manuscript.
Therapeutics Research Initiative Grant from Chiba University School of Medicine (2019-S9 to T.F.) , and Global and Prominent Research Grant from Chiba University (2018-Y9 to A.K) .
A.K. is a member of the Editorial Board of the Chiba Medical Journal. The other authors declare that there is no conflict of interest to disclose.
Not applicable.
Not applicable.
The authors thank all the laboratory members and collaborators who supported the study.
Address correspondence to Dr. Atsushi Kaneda.
Department of Molecular Oncology, Graduate School of Medicine,
Chiba University, 1-8-1, Inohana, Chuou-ku, Chiba 260-8670, Japan.
Phone/Fax: +81-43-226-2039.
E-mail:kaneda@chiba-u.jp
