




Chiba Medical J. 99E:9-16, 2023
doi:10.20776/S03035476-99E-2-P9
〔 Chiba Medical Society Award Review 〕
Yosuke Kurashima1-6)
1) Department of Innovative Medicine, Graduate School of Medicine, Chiba University, Chiba 260-8670.
2) Institute for Advanced Academic Research, Chiba University, Chiba 263-8522.
3) Research Institute of Disaster Medicine, Chiba University, Chiba 260-8670.
4) Synergy Institute for Futuristic Mucosal Vaccine Research and Development, Chiba University (cSIMVa), Chiba 263-8522.
5) International Vaccine Design Center, The Institute of Medical Science, The University of Tokyo, Tokyo 108-8639.
6) Department of Medicine, CU-UCSD Center for Mucosal Immunology, Allergy and Vaccines (cMAV), School of Medicine, UCSD.
(Received January 9, 2023, Accepted February 22, 2023, Published April 10, 2023.)
The imbalance of bacterial communities, known as “dysbiosis”, is thought to be not only correlated with but also the cause of diseases. Recently, it has been considered that the colonization of intestinal bacteria into normally sterile host tissues or extra-intestinal compartments, known as bacterial translocation, causes tissue damage and induces the development of inflammatory diseases. Inflammatory bowel disease (IBD) is linked to bacterial translocation that exacerbate intestinal inflammation by promoting excessive host immune responses or impairing the mucosal barrier. These potentially translocated bacteria are known as pathobionts, defined as symbionts that can promote pathology only when specific genetic, immunological, or environmental conditions are altered in the host. In this review, I herein discuss the current understanding of the host mucosal defense, specifically with regard to the pancreas–intestinal barrier axis, for the control of pathobionts in intestinal inflammation.
Pancreas-gut axis, Glycoprotein 2, Inflammatory bowel diseases, Extra-intestinal manifestation
The imbalance of bacterial communities, known as “dysbiosis”, is considered correlated with disease onset; however, accumulated evidence has revealed that dysbiosis is also the cause of diseases[1]. Some commensal microorganisms, or “symbionts”, are classified as ‘pathobionts’. Pathobionts are organisms that can cause harmful responses, becoming “pathogenic” in the wake of the breakdown of the delicately balanced homeostatic condition between the host immune system and commensal microbiota[2-5].
Regarding the etiology of Crohn’s disease (CD) and ulcerative colitis (UC), it is generally accepted that a dysregulated immune response against commensal bacteria triggers the pathogenesis of gut inflammation[6,7]. The gut chronic inflammation associated with CD causes relapsing tissue damage as well as strictures and fibrosis. CD-associated stricture and fibrosis are frequently unresponsive to anti-inflammatory agents[8]; therefore, there is a need to identify additional targets for novel preventive and therapeutic strategies. In addition, between 25% and 40% of IBD patients experience extraintestinal manifestations or complications as the disease affects other parts of the body, commonly including the joints, skin, bones, eyes, kidney, and liver[9,10], and the translocation of pathobionts from the luminal side is a causative event for systemic symptoms[5].
Bacterial translocation is defined as the passage of pathogens and commensal bacteria from the gastrointestinal tract to extraintestinal sterile tissues, such as mesenteric lymph nodes, liver, spleen, and kidney[2-5,11,12]. IBD is linked to bacterial translocation that exacerbates intestinal inflammation by promoting excessive host immune responses or impairing the mucosal barrier[13-20]. In addition to pathogenic bacteria, the invasion of indigenous gut commensal bacteria through the intestinal mucosa into the tissue eventually causes diseases via the aggravation of local and systemic inflammation[21-23]. For example, adherent invasive Escherichia coli (AIEC) has been shown to form a biofilm at the epithelial cell layer and induces not only epithelial IL-8 production but also macrophages to produce TNF, which exacerbates inflammation[24,25]. Enteococcus spp., such as Enterococcus faecalis and Enterococcus hirae found in the mesenteric fat, omentum, mesenteric lymph nodes, and spleen, also induce inflammatory responses[22,26]. In addition, Proteus spp. Morganella, and Providencia spp. can cause colitis by producing urease[27-29], while Mucispirillum can exacerbate gut inflammation by driving colitogenic Th1 CD4+ T cells[30].
These potentially translocated bacteria, known as pathobionts, can be considered targets of disease onset; therefore, understanding the host defensive response to these pathobionts is important[4,31].
The human body is equipped with several layers of mucosal defense system against pathobionts. Antimicrobial peptides are secreted by Paneth cells located in the small intestinal crypts of Lieberkühn and highly specialized secretory epithelial cells[32]. Defensins, also produced by Paneth cells, balance the microbiota composition and protect the host from invading bacteria. Mucus produced by goblet cells plays a key role in the maintenance of the necessary spatial hostmicrobial segregation[33]. In addition, secretory IgA plays a pivotal role in neutralizing bacterial exotoxins and antigens. The induction of IgA to Clostridium ramosum, promoting high-fat-induced obesity by enhancing nutrient absorption, can ameliorate obesity and other metabolic consequences[34]. It is also known that some pathobionts escape clearance by inducing immune tolerance against themselves[35]. Thus, it is also important to understand the host mucosal immune system against pathobionts.
In addition to the mucosal immune barrier system produced by intestinal epithelium and immune cells against pathobionts[36], the pancreas secretes several kinds of proteins, such as regenerating gene (Reg)[37], secretory phospholipase A2 (sPLA2)[38], pancreatic lipase[39], and trypsin[40]. These proteins protect the gut through anti-microbial effects by inducing bacterial lysis, protecting the epithelial layer[37], balancing intestinal microbiota[39], and activating other germicidal proteins (e.g. Reg3α/γ, α-defensin)[41,42].
Our group recently revealed the importance of the pancreas-gut organ axis for the protection against bacterial translocation via the secretion of GP2 from pancreatic acinar cells, which act as a first line of defense against adhesive and invasive pathobionts, such as AIEC[43,44].
As mentioned above, AIEC binds to epithelial cells by adhering to mannose residues expressed on CEACAM6 and TLR4 via the type 1 pili adhesin FimH[45]. FimH is essential for adhesion and triggering inflammation. FimH binding to TLR4 induces the production of inflammatory cytokines, such as TNF, IL- 6, and IL-8[46]. FimH-mediated bacterial infiltration and inflammatory responses are thus considered novel targets for IBD treatment[47,48].
GP2, an FimH-binding molecule, is a glycosylphosphatidylinositol (GPI) -anchored protein that was first reported in the pancreas[49] and is the most abundant and major membrane protein found in zymogen granules of exocrine pancreas acinar cells[50,51]. GP2 is abundantly secreted into the duodenum along with the pancreatic juice, of which more than 2 L is released daily[52] (Fig. 1).
Levels of IgA and IgG against GP2 are increased in the serum and intestinal lumen of IBD patients, and these neutralizing anto-antibodies are considered to be involved in the pathogenesis of intestinal inflammation[53,54]. We assessed the location of GP2 in the gastrointestinal tract and found that GP2 is diffusely distributed throughout the intestinal tract, from the duodenum to the colon[43]. GP2 is known to be expressed on the surface of M cells, which are specialized cells found in the follicle-associated epithelium of intestinal Peyer’s patches[51,55]. We therefore investigated mice lacking GP2 specific to pancreatic acinar cells and found that luminal GP2 was completely diminished in these mice[43] (Fig. 2). These results indicate that luminal GP2 is totally derived from the pancreas.
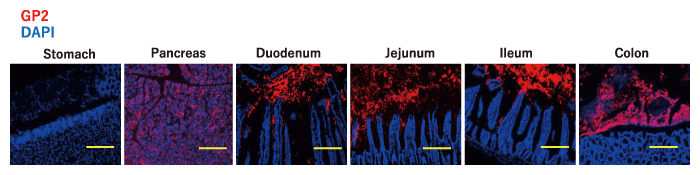
Fig. 1 Influx of pancreatic GP2 into the intestinal lumen. The stomach, pancreas, and each part of the intestine of mice were stained with GP2 (red) and DAPI (blue). Scale bar: 100 μm. Modified from[43].
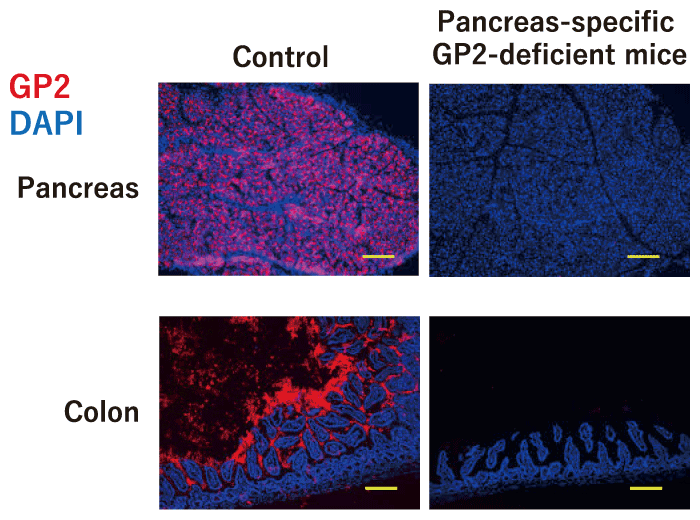
Fig. 2 Complete disappearance of intestinal luminal GP2 in pancreas-specific GP2-deficient mice. The pancreas and intestine of pancreatic acinar cell-specific GP2-deficient mice and control mice were stained with GP2 (red) and DAPI (blue). Scale bar: 100 μm. Modified from[43].
It has been reported that there is no marked difference in the pancreatic function or morphology of mice lacking GP2 compared with normal mice, so the precise role of pancreatic GP2 remains unclear[56]. Tamm-Horsfall protein (THP), a homologue of GP2, is the principal urinary protein that binds specifically to type 1 fimbriated E. coli, the main cause of urinary tract infection[57]. THP suppresses the adhesion of E. coli to epithelial cells. A physiological concentration of THP completely abolished the binding of E. coli to its receptors, uroplakins Ia and Ib, and protected against urinary inflammation[58]. Thus, like THP, GP2 may play important roles in preventing pathogenic entities and pathobionts from binding to epithelial cells. Indeed, in co-culture with GP2 and E. coli, GP2 binds to E. coli in an FimH-dependent manner. In addition, histochemical and flowcytometry analyses have revealed the co-localization and adhesion of GP2 to the bacteria. In particular, about 5% of bacteria in feces bind to GP2 in mice.
To clarify the roles of pancreatic GP2, a dextran sodium sulfate (DSS) -induced colitis model was examined, and inflammation was found to be more severe under conditions of GP2 deficiency than GP2 sufficiency[43]. In addition, serum auto-antibodies to E. coli were found in GP2-deficient colitis mice. These results collectively indicated that commensal bacteria pass through the mucosa and infiltrate systemic compartments, inducing systemic immune responses. Inoculation of bacteria associated with GP2 to the ligated colonic loop revealed the inhibition of bacterial infiltration in the mucosa by GP2 association[43]. These results collectively indicated that pancreatic GP2 plays a pivotal role in controlling microbiome translocation and inducing mucosal protection[43].
Inflammation, accompanied by damage of epithelial integrity, reduces the production of defensin and mucin and eases bacterial translocation. In contrast, GP2 levels in the luminal contents are increased in colitic mice with severe intestinal inflammation, and its secretion from acinar cells is increased in colitic mice as well[43].
The underlying mechanisms of GP2 release during inflammation were evaluated by an in vitro stimulation experiment involving inflammatory cytokines (e.g. TNF and IL-6) and PAMPs (e.g. lipopolysaccharide) with the stimulation of pancreatic acinar cells, and TNF was the only cytokine found to enhance GP2 production[43].
The administration of TNF to the intraperitoneal compartment of mice increased the production of GP2. Simultaneously, the administration of an anti-TNF neutralizing antibody to colitic mice suppressed GP2 enhancement. Taken together, these results indicated that inflammatory crosstalk between pancreas and intestine mediates mucosal protection.
During inflammation, extraintestinal tissues and organs to the intestine, including pancreas, sense inflammatory signals and enhance the production of GP2 for the regulation of the microbiome. The homeostatic machinery underlying GP2 release has also been gradually revealed by our group. It is important to further understand the pancreatic-gut axis to develop novel approaches for the prevention and treatment of diseases.
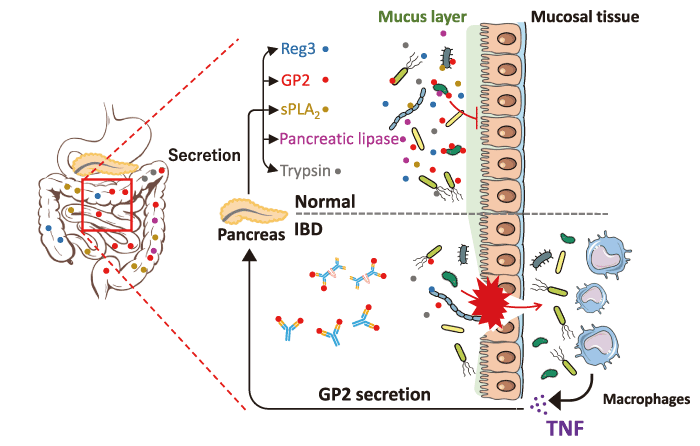
Fig. 3 The pancreatic-gut axis for the regulation of the microbiome. GP2, Reg, sPLA2, lipase, and trypsin are pancreatic secretory components regulating the pathobionts. These agents protect the epithelial barrier and activate anti-microbial peptides, playing a pivotal role in mucosal defense. Anti-GP2 auto-antibodies produced in IBD patients neutralize the activities of GP2 in the microbiome. Translocated bacteria stimulate immune cells, such as macrophages, causing inflammatory cytokine release. TNF enhances the expression of GP2 under inflammatory conditions.
GP2, Reg, sPLA2, lipase, and trypsin are pancreatic secretory components against the microbiome. These agents protect the epithelial barrier and activate antimicrobial peptides, playing a pivotal role in mucosal defense[37,39,59,60]. The effects of these agents against bacteria are not fully understood. As mentioned above, bacterial translocation induces not only exacerbation of intestinal inflammation but also multiorgan dysfunction. Thus, it is important to reveal and target the pancreatic-gut axis for the regulation of IBD and extra-intestinal manifestations.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants from The Ministry of Education, Culture, Sports, Science, and Technology (MEXT) for LEADER; Japan Agency for Medical Research and Development (AMED) PRIME (20gm6010012h0004/20gm6210024h0001) and Project Focused on Developing Key Technology for Discovering and Manufacturing Drugs for Next- Generation Treatment and Diagnosis, The nextgeneration drug discovery and development technology on regulating intestinal microbiome (NeDDTrim) (JP21ae0121040) ; Japan Society for the Promotion of Science (JSPS) for Grant-in-Aid for Scientific Research S (18H05280) and Scientific Research B (19H03450), Challenging Research (Exploratory) 21K19494], Funds for the Promotion of Joint International Research (18KK0432), Future Medicine Funds at Chiba University, Danone Institute of Japan Foundation, The Naito Foundation, Hoyu Science Foundation, Waksman foundation of Japan, Yamada Science Foundation, and the Chiba University-UC San Diego Center for Mucosal Immunology, Allergy, and Vaccines (cMAV).
Not applicable.
Not applicable.
I would like to thank to all collaborators and lab research members for supporting the project. I would also like to express my sincere gratitude to Distinguished Professor. Hiroshi Kiyono (Chiba University) for guiding the research.
Address correspondence to Dr. Yosuke Kurashima.
Department of Innovative Medicine, Graduate School of Medicine,
Chiba University, 1-8-1, Inohana Chuou-ku, Chibashi,
Chiba, 260-8670, Japan.
Phone: +81-43-226-2848.
Fax: +81-43-226-2183.
E-mail: yosukek@chiba-u.jp
