




Chiba Medical J. 91E:1~6,2015
doi:10.20776/S03035476-91E-1-P1
[The Chiba Medical Society Award (2014) ]
Yoshihiro Onouchi
Department of Public Health, Graduate School of Medicine, Chiba University, Chiba 260-8670.
( Accepted November 29, 2014 )
Kawasaki disease (KD) is a systemic vasculitis of unknown etiology that mainly affects infants and children. To unravel the genetic background of KD, we have been searching for relevant susceptibility genes. As the first step, we performed a genome-wide linkage study and identified 10 candidate chromosomal regions. In a subsequent case-control association study of single nucleotide polymorphisms (SNPs) mapped within the candidate regions, we identified functional SNPs of ITPKC and CASP3 that were significantly associated with KD. In a genome-wide association study of KD, we then identified the SNPs conferring KD susceptibility in the FAM167A-BLK, CD40, and HLA class II gene regions. We also confirmed the previously reported association of a functional SNP of FCGR2A. These findings have provided further insight into the pathogenesis of KD. In particular, by identifying a synergistic association of the susceptibility alleles of ITPKC and CASP3 which confer risks for intravenous immunoglobulin resistance and coronary artery lesion formation, our group highlighted the importance of Ca2+/NFAT pathway activation in KD pathogenesis. On this theoretical basis, we support the administration of cyclosporine, a drug that targets this pathway, in cases of refractory KD.
Kawasaki disease, genetic, genome-wide studies
Kawasaki disease (KD; MIM 611775) , also known as mucocutaneous lymph node syndrome, was first described in 1967 by Dr. Tomisaku Kawasaki, a graduate of Chiba University[1]. KD is a systemic vasculitis that predominantly affects infants and children under 5 years of age is characterized by high fever, polymorphous skin rash, bilateral conjunctivitis, palmar/plantar erythema followed by membranous desquamation, redness of the oral mucosa and lips, and non-suppurated cervical lymphadenopathy. KD was initially thought to be a benign disorder due to its selflimiting nature in many cases. However, an investigation into rare cases of sudden death in KD revealed that most died due to rupture or occlusion of giant coronary artery aneurysms; in fact, it was found that 20-25% of KD patients develop coronary artery aneurysm or dilation if they do not receive adequate treatment in the acute phase [2]. It is these cardiac sequelae that have made KD a leading cause of acquired heart disease in developed countries. Previous nationwide outbreaks experienced in 1979, 1982, and 1986 in Japan and seasonal changes in the incidence rate indicate the involvement of an infectious agent in the etiology of KD. However, despite intensive research, the etiological agent responsible for triggering the disease has not been identified. In the meantime, epidemiological studies have highlighted a marked difference in disease prevalence among ethnicities. The most recent worldwide annual incidence rates per 100,000 children aged 0-4 years was highest in Japan (264.8) [3], followed in order by Korea (134.4) and Taiwan (66.2) [4,5]. Familial aggregation of KD is also known to occur [6-8]. Thus KD has been recognized as a multifactorial disease, the etiology of which involves both environmental and genetic factors. To unravel the genetic background of KD, we have been supporting genome-wide searches for KD susceptibility genes.
Because candidate gene study was thought to be less efficient for complex disorders, especially when information about their pathophysiology is limited, we adopted a genome-wide approach in which we made no assumptions about the relevance of candidate genes and considered all genes in the human genome as candidates. As the first step in our research, we conducted a genome-wide linkage study in efforts to map susceptibility genes to chromosomes. Using an affected sib-pair method, we assessed allele sharing of genetic markers among sibling pairs in whom both were affected with KD, and searched for markers with alleles shared between them at an above-expected rate. In this analysis, excess allele sharing indicates linkage between the marker and the disease gene. A total of 399 microsatellite markers selected at an average interval of 9.9 cM from 22 autosomes and the X chromosome were genotyped for 78 affected sib-pairs with KD and their family members. Ten chromosomal regions (4q35, 5q34, 6q27, 7p15, 8q24, 12q24, 18q23, 19q13, Xp22, and Xq27; Figure 1) showing a nominal trend of linkage (MLS > 1.0) were identified [9]. This was the first successful genome-wide study for KD and led to the identification of 2 susceptibility genes.
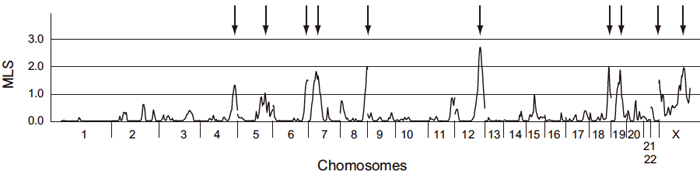
Fig.1 Results of sibling pair linkage analysis.
Ten chromosomal regions have positive linkage signals (MLS>1.0). Chromosome numbers 1-22 and X are shown on the X-axis.
We continued narrowing down the candidate regions identified in the linkage study. Using information from the SNP map constructed in Japan as part of the Japanese Millennium Genome Project, we selected SNPs near each linkage peak and compared allele frequencies between KD cases and controls. In this linkage disequilibrium mapping, we found multiple SNPs in the 19q13.2 region that showed replicable association with KD[10]. Through studies of gene and SNP function, we concluded that the inositol 1,4,5-trisphosphate 3-kinase C gene (ITPKC) was the susceptibility gene of the locus and that the rs28493229 SNP was responsible for this susceptibility. We found that the amount of ITPKC transcripts from the associated allele (C) of the SNP was reduced in peripheral blood mononuclear cells (PBMCs) compared with that from the opposite allele (G), and that this difference was due to change in the splicing efficiency of intron 1 of the gene. The rs28493229 SNP is located at the 9th nucleotide position of ITPKC intron 1, and we found that the transcripts from the C allele may persist unspliced until being degraded by the nonsense-mediated decay mechanism. ITPKC is a kinase of inositol 1,4,5-trisphosphate (IP3), which is generated by hydrolysis of phosphatidylinositol 4,5-bisphosphate in response to stimulation of various cell surface receptors such as T-cell, B-cell, Fc gamma, and G-protein-coupled receptors. IP3 acts as a second messenger molecule in downstream signal transduction. ITPKC might therefore downregulate the Ca2+/NFAT pathway by phosphorylating IP3 into IP4, which does not bind to the IP3 receptor, consequently preventing signal transduction (Figure 2).
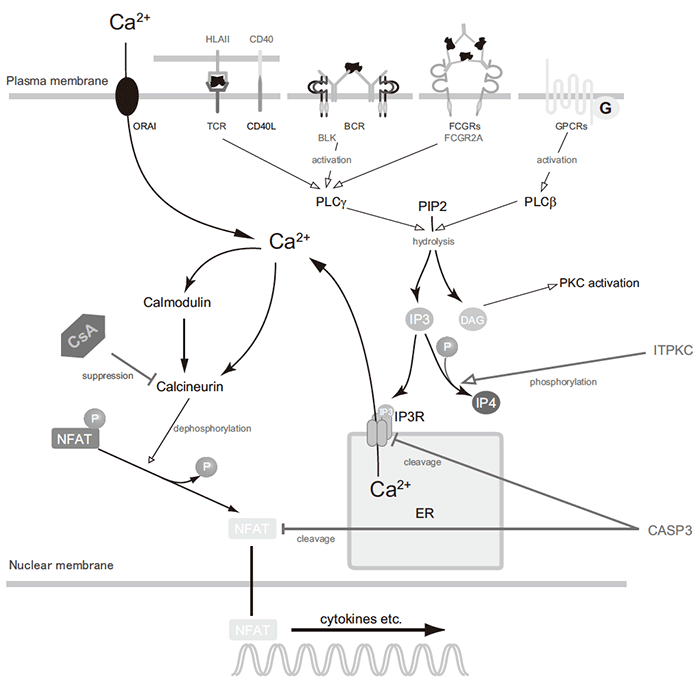
Fig.2 Possible roles of KD susceptibility genes in the Ca2+/NFAT pathway.
BCR:B-cellreceptor; CASP3:caspase-3; CsA: cyclosporine A; DAG: diacylglycerol; ER: endoplasmic reticulum; FCGRs: Fc gamma receptors; GPCRs: G-protein-coupled receptors; IP3: inositol 1,4,5-trisphosphate; IP3R: inositol 1,4,5-trisphosphate receptor; IP4: inositol 1,3,4,5-tetrakisphosphate; ITPKC: inositol 1,4,5-trisphosphate 3-kinase C; NFAT: nuclear factor of activated T-cells; PIP2: phosphatidylinositol 4,5-bisphosphate; PKC: protein kinase C; PLC: phospholipase C; TCR: T-cell receptor.
By applying positional candidate gene study to the 4q35 region, we identified SNPs associated with KD around caspase-3 (CASP3). Sequences around rs113420705, one of the associated SNPs and located in exon 1 of CASP3, has enhancer activity related to nuclear factor of activated T-cells (NFAT); owing to alteration of the activity, the risk allele (A) expresses smaller amount of CASP3 mRNA in PBMCs than the opposite allele (G) [11]. As CASP3 is known to play a pivotal role in immune cell apoptosis, reduced CASP3 expression is thought to allow sustained activation of immune cells and progression of KD inflammation. Association of both ITPKC and CASP3 SNPs with KD susceptibility has gained credibility after validation in multiple ethnicities [12-14].
Development of high throughput genotyping platforms for SNPs has made genome-wide scans of susceptibility genes of complex diseases a reality. In particular, establishment of the genome-wide association study( GWAS) method has dramatically promoted these studies. Today, 9 GWAS papers for KD have been published and a number of genomic regions have been proposed in one or more papers as susceptibility loci for KD. However, many of these regions failed to satisfy the formal significance thresholds used in genomewide studies and therefore required further follow up or validation. Along with ITPKC and CASP3 genes, associations of SNPs in the FCGR2A, BLK, and CD40 gene regions identified in GWAS to date have been validated in multiple ethnic groups and are generally considered to be credible susceptibility genes or loci for KD. Through our work, we have contributed to the identification of association signals in BLK and CD40 gene regions. In our GWAS, significant and reproducible association of the SNPs in the HLA class II gene region was also identified.
In 2011, an international consortium for the genetic study of KD organized by Dutch, British, Australian, and American researchers reported results of a GWAS conducted among European KD patients [13]. In this study, SNPs near the Fc gamma receptor (FCGR) gene cluster located on the chromosome 1q23 region were significantly associated with KD. Association of a functional SNP of Fc fragment of IgG, low affinity IIa, receptor (FCGR2A) gene (rs1801274 A/G) was the most significant and the A allele confers KD susceptibility. FCGR2A expressed on neutrophils and macrophages transduces the activation signal when ligated with immune complexes and clustered on the cell surface. The A-to-G substitution results in a change of translation of the 131st amino acid from histidine to arginine; the histidine type corresponding to the associated allele (A) has higher binding affinity to the IgG2 subclass. We validated the association with this SNP in our GWAS samples and also found that the A allele was overrepresented in Japanese KD patients [15].
In two independent GWAS for KD, conducted by our group [15] and a Taiwanese group [16] , significant associations of SNPs located in chromosome 8p23-p22 region were observed. The association in this locus was highest at the intergenic region between the family with sequence similarity 167, member A (FAM167A) and B lymphoid kinase (BLK) genes. SNPs in this area have been associated with multiple autoimmune diseases. BLK is expressed mainly in B cells and is involved in B-cell receptor signaling. Because B calls play a central role in autoimmunity, BLK rather than FAM167A, which has not been functionally characterized, is generally taken to be a susceptibility gene in autoimmune diseases.
CD40, also known as TNF receptor superfamily member 5, is located on chromosome 20q12-q13.2. CD40, which is expressed on the cell surface of antigen presenting cells and vascular endothelial cells among others, is stimulated when ligated with CD40L, its counterpart expressed on activated CD4 T cells and platelets, and it transduces activation or differentiation signals into the cells. Significant association of the SNPs around CD40 with KD susceptibility was reported in the two GWA studies mentioned above [15,16]. The associated SNPs were in linkage disequilibrium with known functional SNP-altering efficiency of CD40 protein translation( rs1883832 C/T), and the C allele, which corresponds to higher CD40 protein expression, is linked with alleles of the SNPs conferring susceptibility to KD in this area. As is the case with BLK, CD40 is a known common susceptibility gene for autoimmune diseases.
SNPs in the HLA class II region also showed a significant association in our GWAS. The association peaked at the intergenic region between HLA-DQB2 and HLA-DOB [15]. Although we confirmed a positive association of the SNPs in this region in an independent case-control set, association signals observed for the SNPs in this region were not significant in GWAS conducted among other ethnicities. It is therefore likely that there is a genetic variant that determines susceptibility to KD in the HLA class II region, at least in the Japanese population. However, further study is needed to judge whether the variation is Japanese-specific or not. Although extended linkage disequilibrium and numerous genes with high sequence homology as well as densely distributed variations in this area make the determination of true susceptibility genes and variants challenging, the finding of this association in the HLA class II region might serve as a clue to elucidate the controversial contribution of HLA to KD pathogenesis.
Pathologically, KD has been defined as a vasculitis that mainly affects small and medium-sized arteries. However, definitive categorization of KD into disease groups has not been made from the viewpoint of underlying pathophysiology. As mentioned above, genome-wide studies have revealed previously unknown commonalities in genetic factors between KD and other inflammatory diseases (Figure 3). However, variants of ITPKC and CASP3 were not previously associated in any GWAS for other diseases. This indicates that the genetic basis of KD is structured by multiple factors: those shared with other diseases as well as those highly specific for KD. Accumulation of knowledge on these factors will undoubtedly help us understand the pathophysiology of the disease and might highlight the applicability of existing therapies for other diseases to KD.
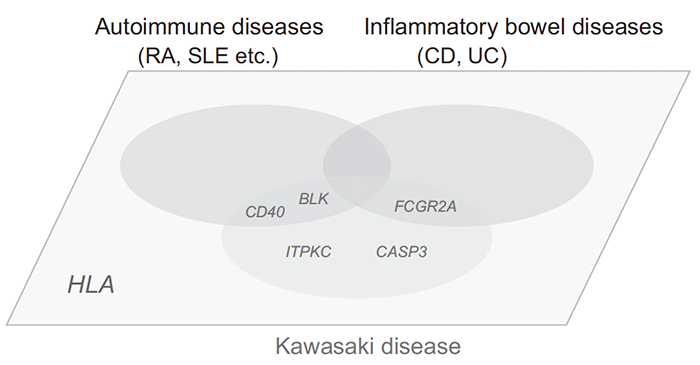
Fig.3 Overlapping of genetic factors for KD and those for other immune or inflammatory diseases unraveled by genome-wide studies.
A combination of oral aspirin and a high dose of intravenous immunoglobulin (IVIG) is standard therapy for patients with acute KD. When administered sufficiently early in the disease course, this treatment promptly leads to defervescence and symptom remission in most patients. However, 10-15% of patients respond poorly, placing them at higher risk of developing coronary artery lesions (ALs). By analyzing KD patients treated with a unified regimen and evaluating their response to this treatment using standardized criteria, we assessed the influence of the susceptibility SNPs of ITPKC and CASP3 on patients' responsiveness to IVIG and risk of developing CAL. In this study, we found that KD patients possessing susceptibility alleles on both ITPKC and CASP3 had increased risk for IVIG resistance and CAL formation [17] . This finding, together with previous knowledge of IP3 receptor, type 1 [18] and NFATc2 [19] cleavage in T cells by CASP3, highlighted the possibility that CASP3 also acts as a negative regulator of the Ca2+/ NFAT pathway in the pathophysiology of KD (Figure 2). Cyclosporine, a calcineurin inhibitor that suppresses this signal transduction pathway specifically, has drawn considerable attention as an effective drug for refractory KD patients [20,21] , and a phase III clinical trial of cyclosporine in KD patients predicted to show resistance to IVIG is currently underway at the initiative of Chiba University (http://www.chiba-crc.jp/kaica_trial-pc/index.html).
Unfortunately, even when their genetic effects are considered together, the currently known susceptibility genes for KD do not fully account for familial aggregation or different incidence ratios among ethnicities. It is likely that there are many more susceptibility genes unidentified as yet. It is also clear that higher statistical power than in previous studies is needed to identify common variants with minor effects that were missed in previous GWAS. However, increasing the sample size is easier said than done, especially for an acute illness where most patients are infants or children. Integration of multiple GWAS is a possible solution to this issue, which can increase the number of samples and consequently statistical power without requiring the collection of new samples or genotyping. We are now conducting a meta-analysis of three GWA studies with Korean and Taiwanese research groups who have published GWAS papers using their own samples.
Recently, the contribution of rare genetic variations and a greater number of individual mutations to the pathogenesis of common diseases has been increasingly recognized. Although the cost is still quite high at present, analyzing thousands of samples using nextgeneration sequencers will become affordable in the near future. Clarifying more and more genetic factors relating to KD, including common, rare, and individual factors, will bring us closer to solving the mystery of this disease.
We sincerely thank all collaborators, KD patients who took part in the study, their families, and medical staff caring for the patients.
Address correspondence to Dr. Yoshihiro Onouchi.
Department of Public Health, Graduate School of Medicine,
Chiba University,
1-8-1 Inohana Chuouku,Chiba 260-8670, Japan.
Phone: +81-43-226-2069. Fax: +81-43-226-2070.
E-mail: onouchy@chiba-u.jp
